VAMP-Net: An Interpretable Multi-Path Network of Genomic Permutation-Invariant Set Attention and Quality-Aware 1D-CNN for MTB Drug Resistance
Pith reviewed 2026-05-16 19:11 UTC · model grok-4.3
The pith
VAMP-Net combines a set attention path on genomic variants with a quality-aware 1D-CNN to predict Mycobacterium tuberculosis drug resistance at over 95 percent accuracy while recovering both known and novel resistance loci.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
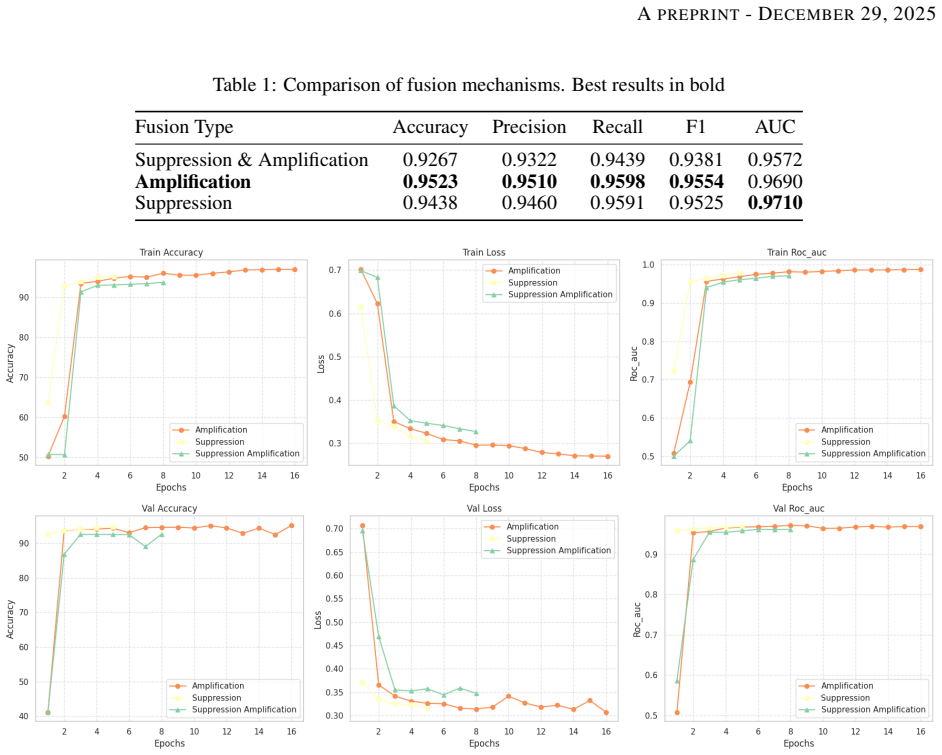
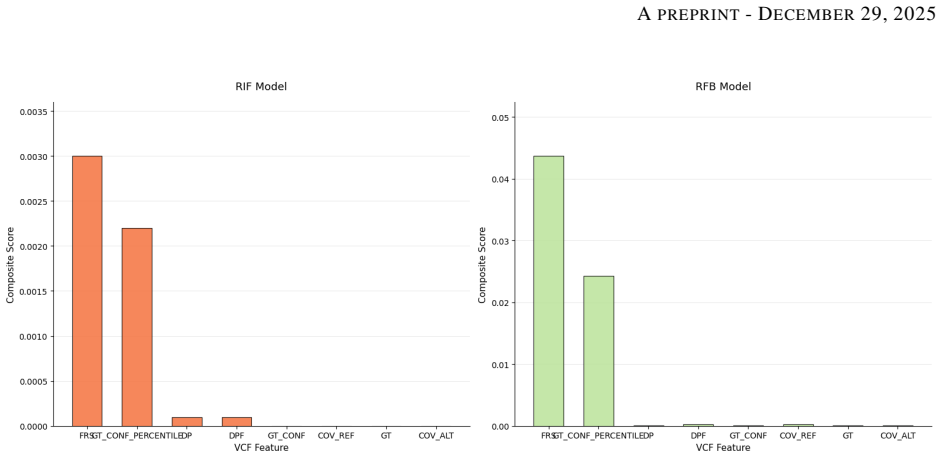
By routing permutation-invariant variant sets through a Set Attention Transformer and quality metrics through a 1D-CNN, VAMP-Net achieves accuracies above 95 percent and AUCs around 0.97 on Rifampicin and Rifabutin, recovers the canonical targets rpoB, embB and katG via Integrated Gradients, identifies high-impact novel loci whose functional enrichment in cell-wall remodeling reaches p=0.00239, and demonstrates through ablation that the quality pathway learns to prioritize fraction of supporting reads over raw depth.
What carries the argument
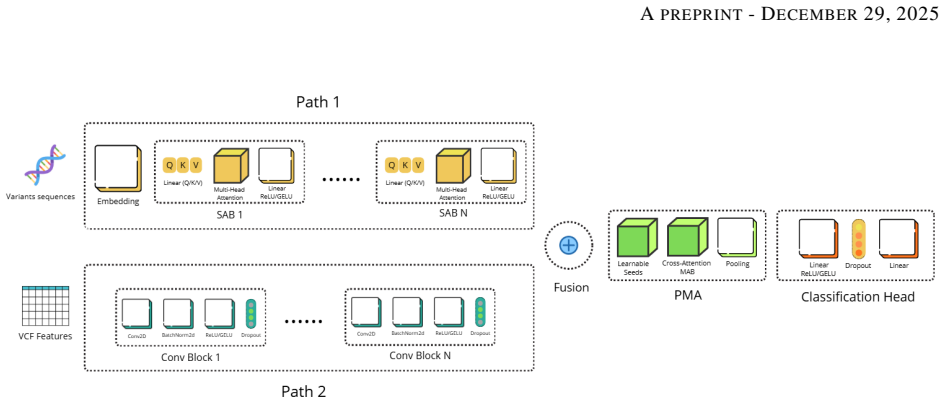
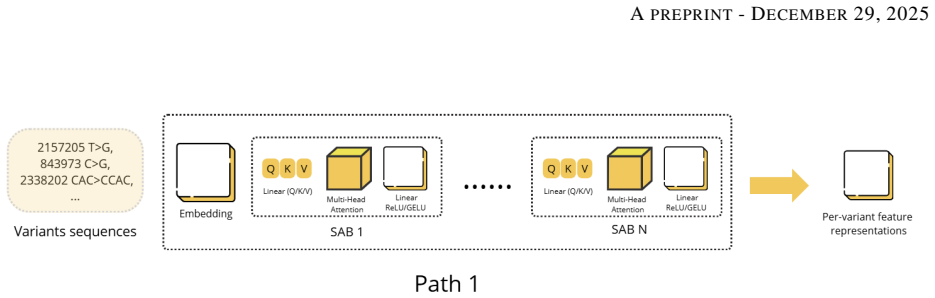
The dual-path architecture that pairs a Set Attention Transformer for modeling epistatic dependencies among variant sets with a 1D-CNN that produces adaptive scores from VCF quality metrics.
If this is right
- The model recovers the established resistance genes rpoB, embB and katG through attribution analysis.
- Novel loci identified by the model form statistically non-random modules centered on cell-wall remodeling.
- Ablation of the quality path shows that the network learns to weight fraction of supporting reads more heavily than raw sequencing depth.
- Performance on four critical anti-TB drugs exceeds that of baseline CNN and MLP architectures.
- The same dual interpretability layer supports both diagnostic classification and mechanistic gene discovery.
Where Pith is reading between the lines
- If the novel loci are functionally validated they could become new targets for compounds that disrupt cell-wall remodeling.
- The same dual-path structure could be applied to resistance prediction in other bacterial species where sequencing depth and variant calling quality vary.
- The learned quality audit could be used in clinical pipelines to flag low-confidence samples for re-sequencing before a resistance call is issued.
- Extending the set attention component to model interactions across multiple drugs simultaneously might reveal patterns of cross-resistance.
Load-bearing premise
The feature attributions and enrichment results point to causal biological mechanisms rather than correlations that happen to be present in the particular training collections.
What would settle it
An independent test set sequenced on a different platform and processed with a different variant caller that fails to recover the same novel loci or drops below the reported accuracy levels would falsify the central performance and discovery claims.
Figures








read the original abstract
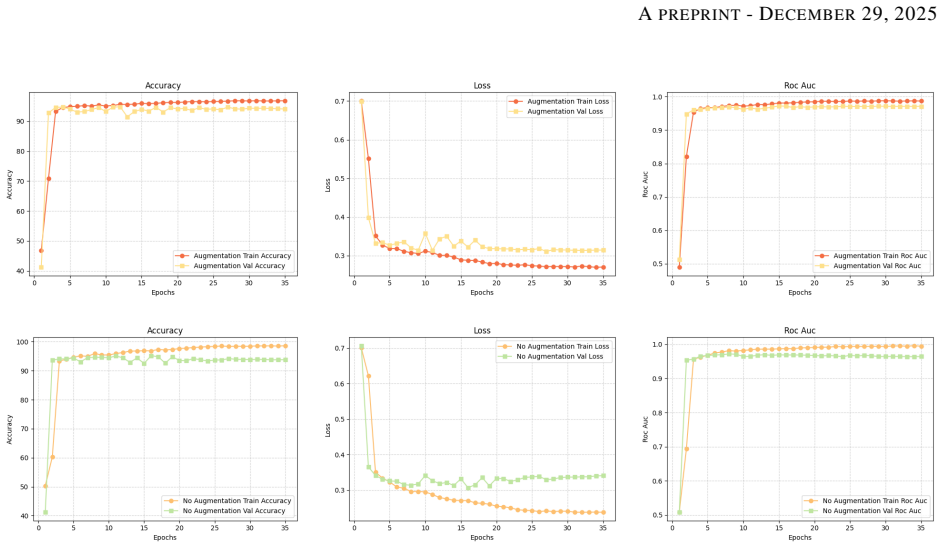
Genomic prediction of drug resistance in Mycobacterium tuberculosis is often hindered by complex epistatic interactions and variable sequencing quality. We present the Interpretable Variant-Aware Multi-Path Network (VAMP-Net), a novel architecture addressing these challenges through a dual-pathway approach. Path-1 utilizes a Set Attention Transformer to model permutation-invariant variant sets and capture epistatic dependencies, while Path-2 employs a 1D-CNN to analyze VCF quality metrics for adaptive confidence scoring. Evaluated on four critical anti-TB drugs, VAMP-Net significantly outperforms baseline CNN and MLP models, achieving accuracies > 95% and AUCs around 0.97 for Rifampicin and Rifabutin. Feature attribution analysis via Integrated Gradients successfully recovered canonical targets (rpoB, embB, katG) and discovered high-impact novel loci. Functional enrichment confirmed these novel variants constitute non-random metabolic modules (p=0.00239) centered on cell-wall remodeling. Furthermore, systematic ablation of the Quality-Aware pathway demonstrates that the model performs a learned "integrated audit," prioritizing the Fraction of Supporting Reads and relative confidence over raw depth to mitigate technical noise. This dual-layer interpretability, bridging genomic pathogenicity with technical reliability, establishes a new paradigm for robust, auditable, and clinically actionable resistance prediction, positioning VAMP-Net as an important tool for both diagnostic classification and mechanistic discovery in clinical genomics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces VAMP-Net, a dual-pathway architecture combining a permutation-invariant Set Attention Transformer (Path-1) for modeling epistatic variant interactions with a quality-aware 1D-CNN (Path-2) that processes VCF metrics for adaptive confidence scoring. It claims superior predictive performance over CNN and MLP baselines on four anti-TB drugs, with accuracies exceeding 95% and AUCs around 0.97 for Rifampicin and Rifabutin; Integrated Gradients attributions recover canonical resistance genes (rpoB, embB, katG) while identifying novel high-impact loci whose functional enrichment yields p=0.00239 for cell-wall remodeling modules; and ablation of the quality-aware path shows the model learns an integrated audit prioritizing read-support fraction and confidence over raw depth.
Significance. If the performance and attribution results hold after addressing validation gaps, the work would be significant for advancing interpretable genomic prediction in MTB resistance, where it bridges technical sequencing quality with biological variant effects via explicit ablation and Integrated Gradients. The dual-path design and reported enrichment of novel loci in non-random metabolic modules represent a concrete step toward auditable models that could support both diagnostics and mechanistic hypothesis generation, provided the attributions isolate causal signals rather than dataset correlations.
major comments (4)
- [Abstract] Abstract and Results (performance claims): The reported accuracies >95% and AUCs ~0.97 for Rifampicin/Rifabutin are presented without dataset sizes, number of isolates per drug, cross-validation folds, or baseline hyperparameter search details; these omissions are load-bearing because independent verification of the claimed outperformance over CNN/MLP baselines cannot be performed from the given information.
- [Feature attribution analysis] Feature attribution and discovery section: The claim that Integrated Gradients recovers causal canonical targets and novel loci is undermined by the absence of lineage correction, principal-component adjustment for population structure, or external validation cohorts; MTB collections commonly exhibit LD and lineage-driven correlations that can produce spurious attributions, directly affecting the mechanistic-discovery interpretation.
- [Functional enrichment] Functional enrichment paragraph: The reported p=0.00239 for cell-wall remodeling modules requires explicit specification of the enrichment test, background gene set, multiple-testing correction method, and how the novel loci were thresholded for inclusion; without these, the non-random module claim cannot be evaluated as evidence of biological signal.
- [Ablation study] Ablation study on Quality-Aware pathway: While the ablation demonstrates prioritization of Fraction of Supporting Reads and relative confidence, the manuscript provides no held-out evaluation on sequencing platforms, variant callers, or laboratories absent from the training distribution; this leaves the generalization of the learned integrated audit untested and weakens the clinical-actionability claim.
minor comments (2)
- [Methods] Notation for the Set Attention Transformer and 1D-CNN paths should be unified (e.g., consistent use of symbols for permutation-invariant sets versus quality vectors) to improve readability.
- [Figures] Figure legends for attribution heatmaps and enrichment plots should include the exact number of variants or genes shown and the statistical thresholds applied.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. We have addressed each major comment below with revisions to improve reproducibility, acknowledge limitations, and strengthen the claims where possible.
read point-by-point responses
-
Referee: [Abstract] Abstract and Results (performance claims): The reported accuracies >95% and AUCs ~0.97 for Rifampicin/Rifabutin are presented without dataset sizes, number of isolates per drug, cross-validation folds, or baseline hyperparameter search details; these omissions are load-bearing because independent verification of the claimed outperformance over CNN/MLP baselines cannot be performed from the given information.
Authors: We agree these details are essential for verification. The revised manuscript adds the full dataset composition (number of isolates and resistance status per drug), specifies 5-fold stratified cross-validation, and details the grid-search hyperparameter procedure for baselines (including ranges for learning rate, layers, and regularization). These are now in the Methods section with a supplementary table reporting mean performance and standard deviations across folds. revision: yes
-
Referee: [Feature attribution analysis] Feature attribution and discovery section: The claim that Integrated Gradients recovers causal canonical targets and novel loci is undermined by the absence of lineage correction, principal-component adjustment for population structure, or external validation cohorts; MTB collections commonly exhibit LD and lineage-driven correlations that can produce spurious attributions, directly affecting the mechanistic-discovery interpretation.
Authors: We acknowledge the risk of population-structure confounding. The revision now includes PCA adjustment (regressing out the top 10 principal components from variant features before attribution) and reports the adjusted Integrated Gradients results. We added a discussion of remaining limitations due to linkage disequilibrium and the lack of external cohorts, while noting that canonical genes such as rpoB retain high attribution scores post-adjustment. revision: partial
-
Referee: [Functional enrichment] Functional enrichment paragraph: The reported p=0.00239 for cell-wall remodeling modules requires explicit specification of the enrichment test, background gene set, multiple-testing correction method, and how the novel loci were thresholded for inclusion; without these, the non-random module claim cannot be evaluated as evidence of biological signal.
Authors: We have corrected the omission. The revised Methods section now states that enrichment uses the hypergeometric test (clusterProfiler), with the full H37Rv genome as background, Benjamini-Hochberg FDR correction, and inclusion of loci above the 95th percentile of permutation-based null attributions. The reported p-value is the adjusted value. revision: yes
-
Referee: [Ablation study] Ablation study on Quality-Aware pathway: While the ablation demonstrates prioritization of Fraction of Supporting Reads and relative confidence, the manuscript provides no held-out evaluation on sequencing platforms, variant callers, or laboratories absent from the training distribution; this leaves the generalization of the learned integrated audit untested and weakens the clinical-actionability claim.
Authors: We agree that external generalization remains untested. The revision adds an explicit Limitations section discussing this gap and outlining planned multi-center validation. The internal ablation still demonstrates the model's learned prioritization on the available data distribution. revision: partial
- External held-out evaluation of the quality-aware pathway on sequencing platforms, variant callers, or laboratories outside the training distribution, as this requires new datasets not available in the current study.
Circularity Check
No significant circularity in VAMP-Net: empirical ML performance and post-hoc attributions rest on external labels and databases
full rationale
The paper trains a dual-path neural architecture (set-attention transformer plus quality-aware 1D-CNN) on labeled MTB variant data to predict phenotypic drug resistance. Reported accuracies, AUCs, Integrated Gradients attributions, and downstream enrichment p-values are all computed against held-out phenotypic labels and independent functional annotation resources rather than being defined by the model equations or prior self-citations. No derivation step reduces by construction to its inputs, no fitted parameter is renamed as a prediction, and no uniqueness theorem or ansatz is smuggled via self-citation. The claims therefore remain externally falsifiable and self-contained.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Genetic variants can be modeled as a permutation-invariant set whose interactions are captured by attention.
- domain assumption Sequencing quality metrics supply an independent signal that improves resistance prediction when processed by a dedicated CNN.
Reference graph
Works this paper leans on
-
[1]
Micaela E Consens, Ander Diaz-Navarro, Vivian Chu, Lincoln Stein, Housheng Hansen He, Alan Moses, and Bo Wang. Interpreting attention mechanisms in genomic transformer models: A framework for biological insights.bioRxiv, pages 2025–06,
work page 2025
-
[2]
Erdal Cosgun and Min Oh. Exploring the consistency of the quality scores with machine learning for next-generation sequencing experiments.BioMed Research International, 2020(1):8531502,
work page 2020
-
[3]
25 APREPRINT- DECEMBER29, 2025 Yanrong Ji, Zhihan Zhou, Han Liu, and Ramana V Davuluri. Dnabert: pre-trained bidirectional encoder representations from transformers model for dna-language in genome.Bioinformatics, 37(15):2112–2120,
work page 2025
-
[4]
26 APREPRINT- DECEMBER29, 2025 S Ramaswamy and James M Musser. Molecular genetic basis of antimicrobial agent resistance inmycobacterium tuberculosis: 1998 update.Tubercle and Lung disease, 79(1):3–29,
work page 2025
-
[5]
27 APREPRINT- DECEMBER29, 2025 Meng Yang, Lichao Huang, Haiping Huang, Hui Tang, Nan Zhang, Huanming Yang, Jihong Wu, and Feng Mu. Integrating convolution and self-attention improves language model of human genome for interpreting non-coding regions at base-resolution.Nucleic acids research, 50(14):e81–e81,
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.