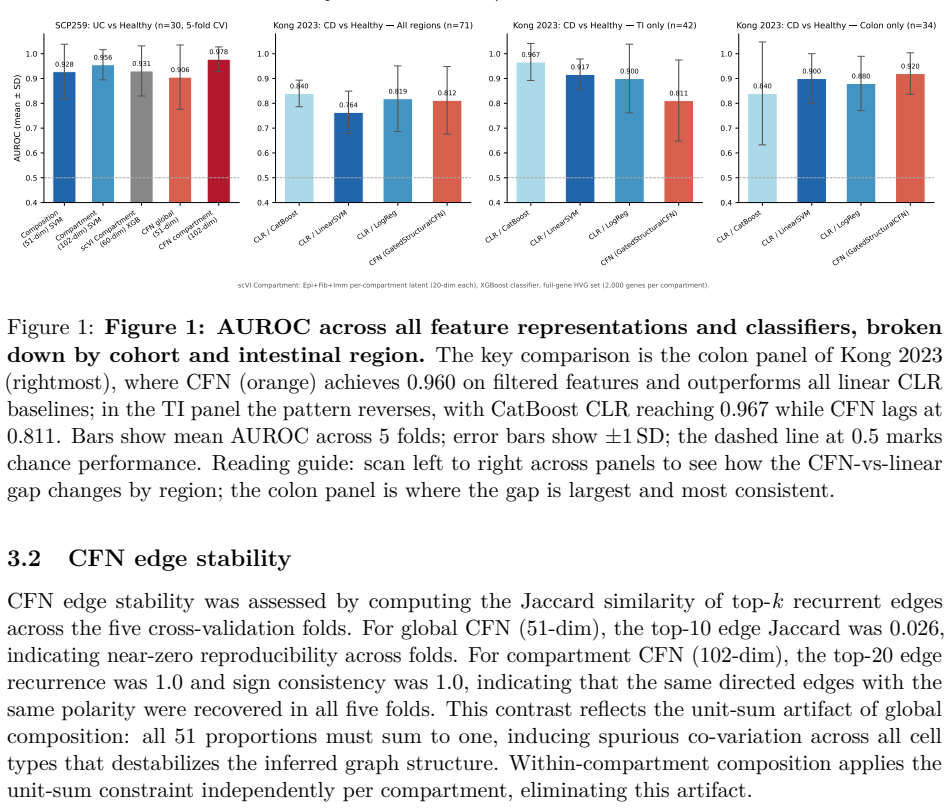
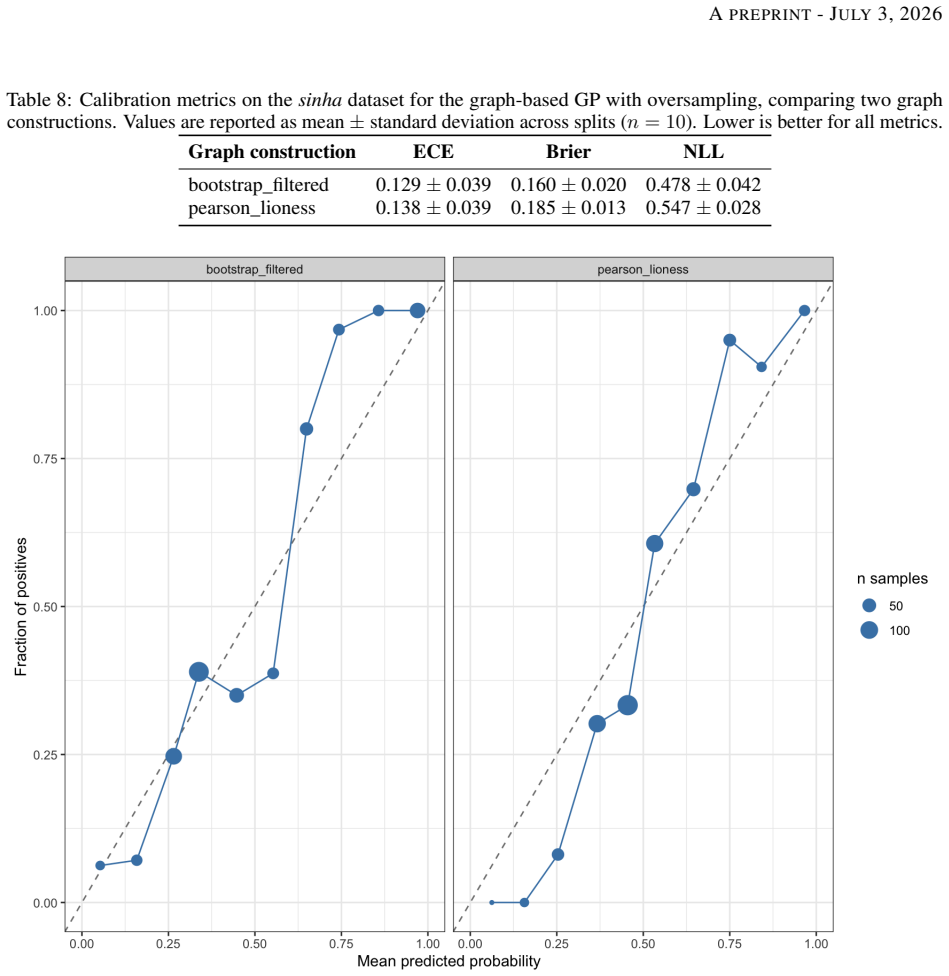
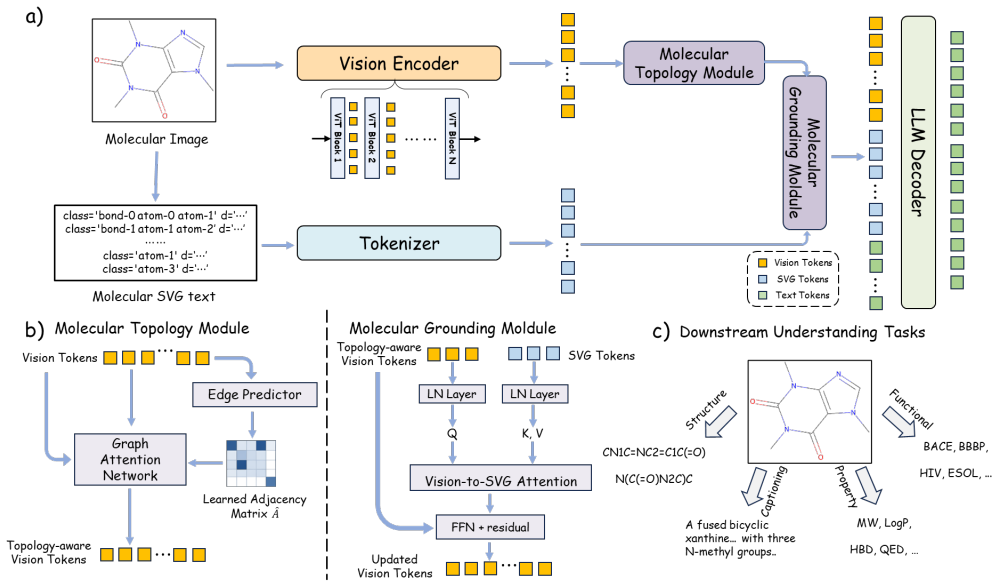
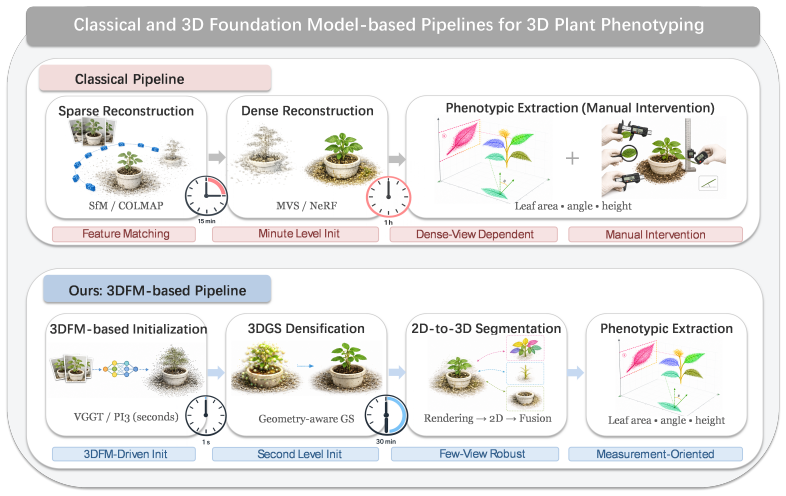
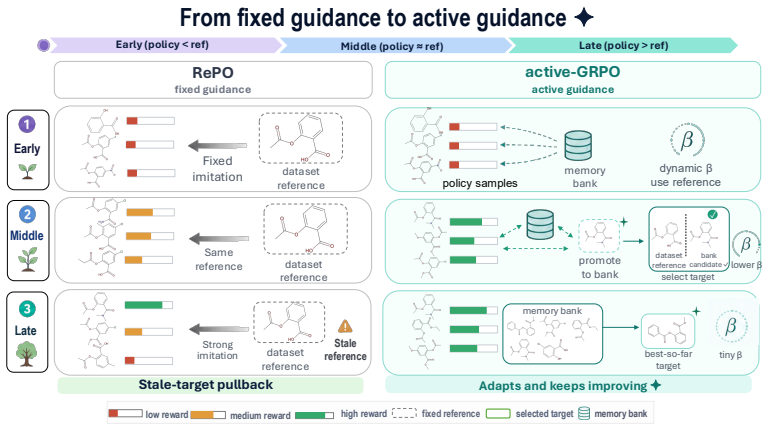
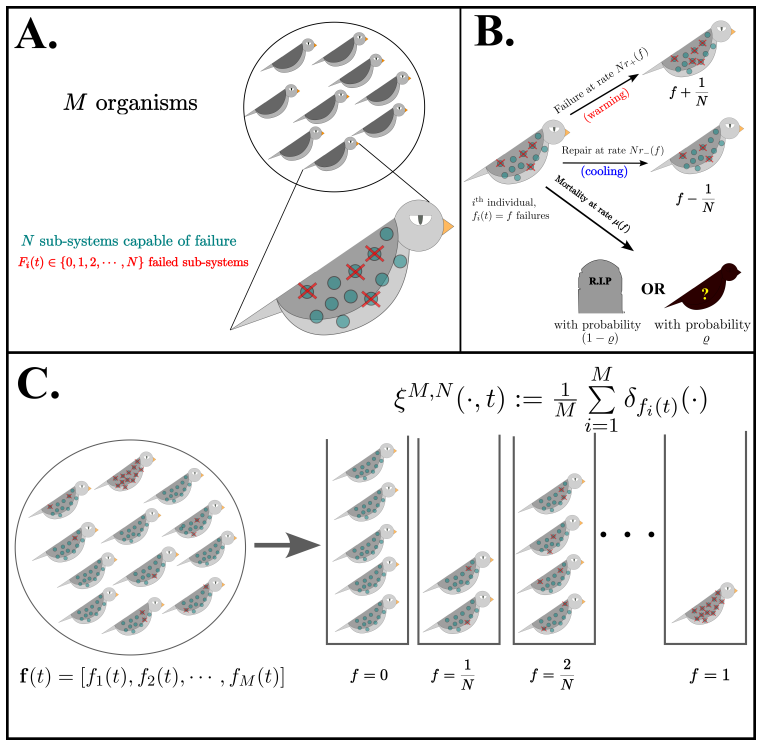
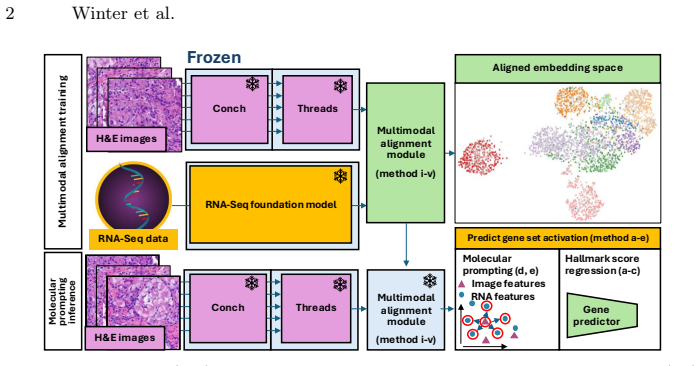
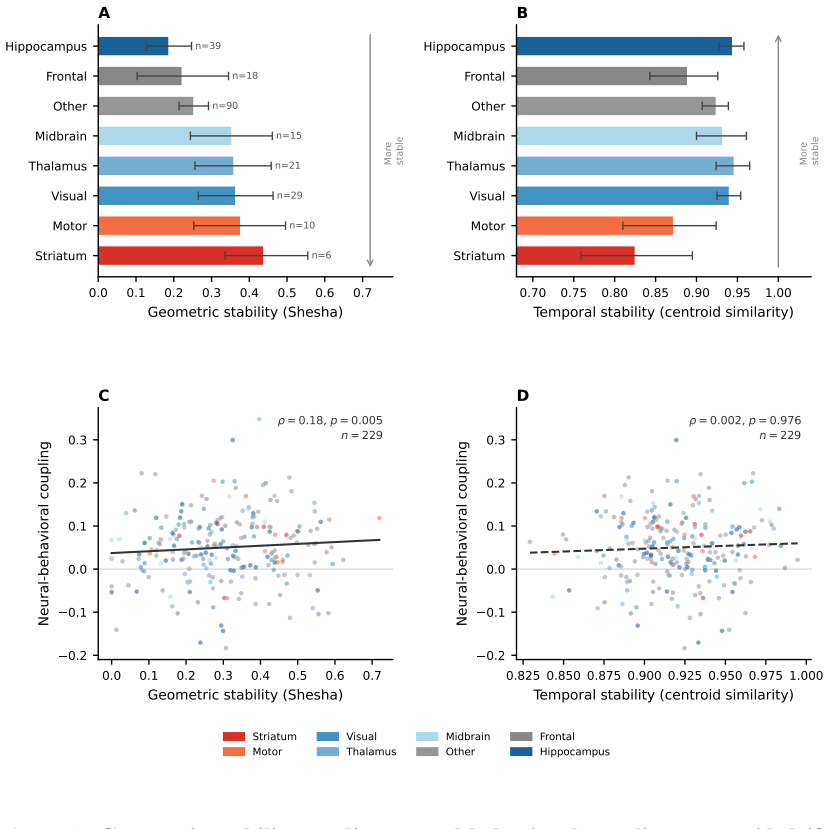
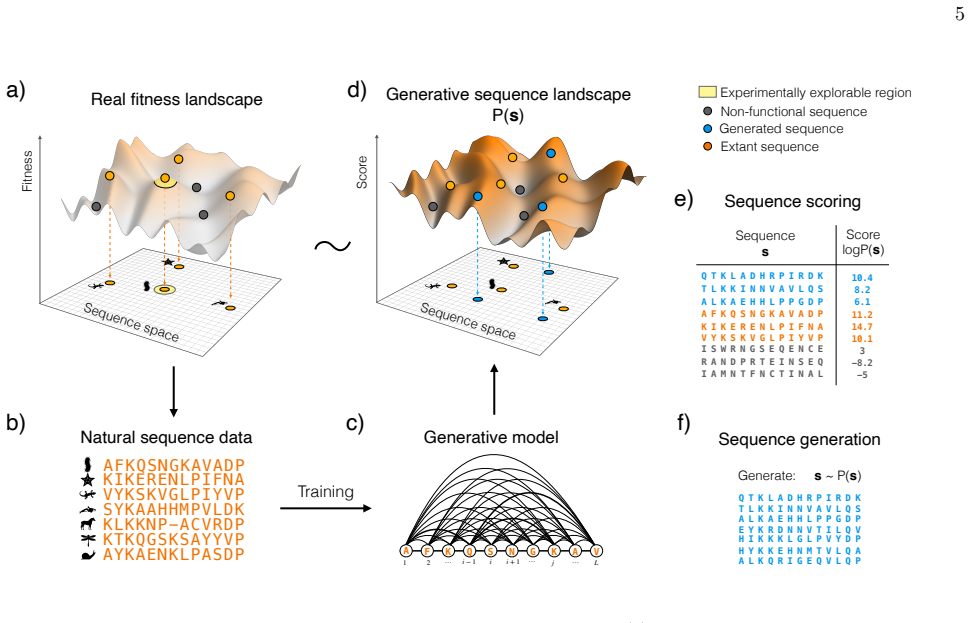
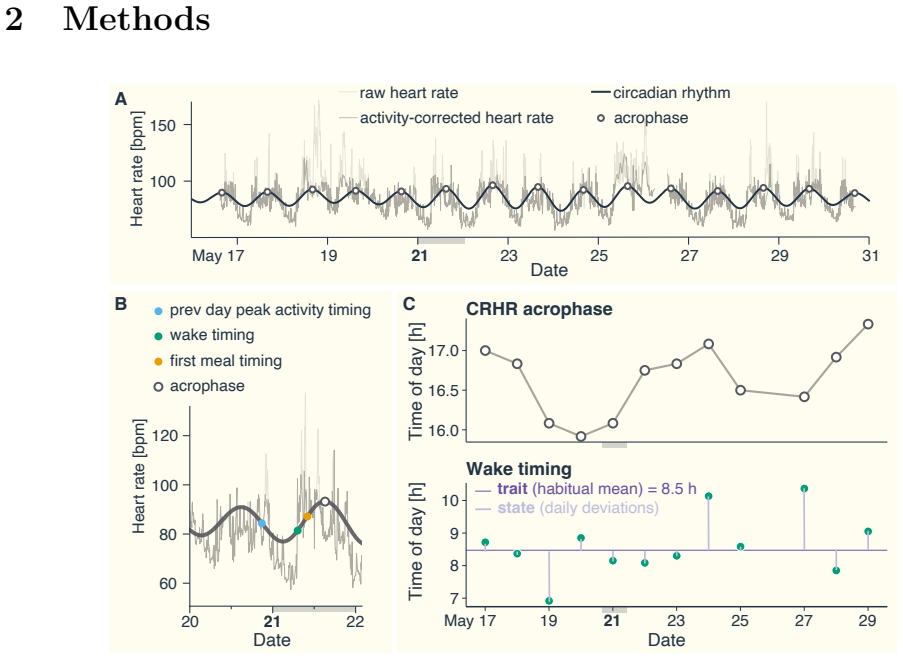
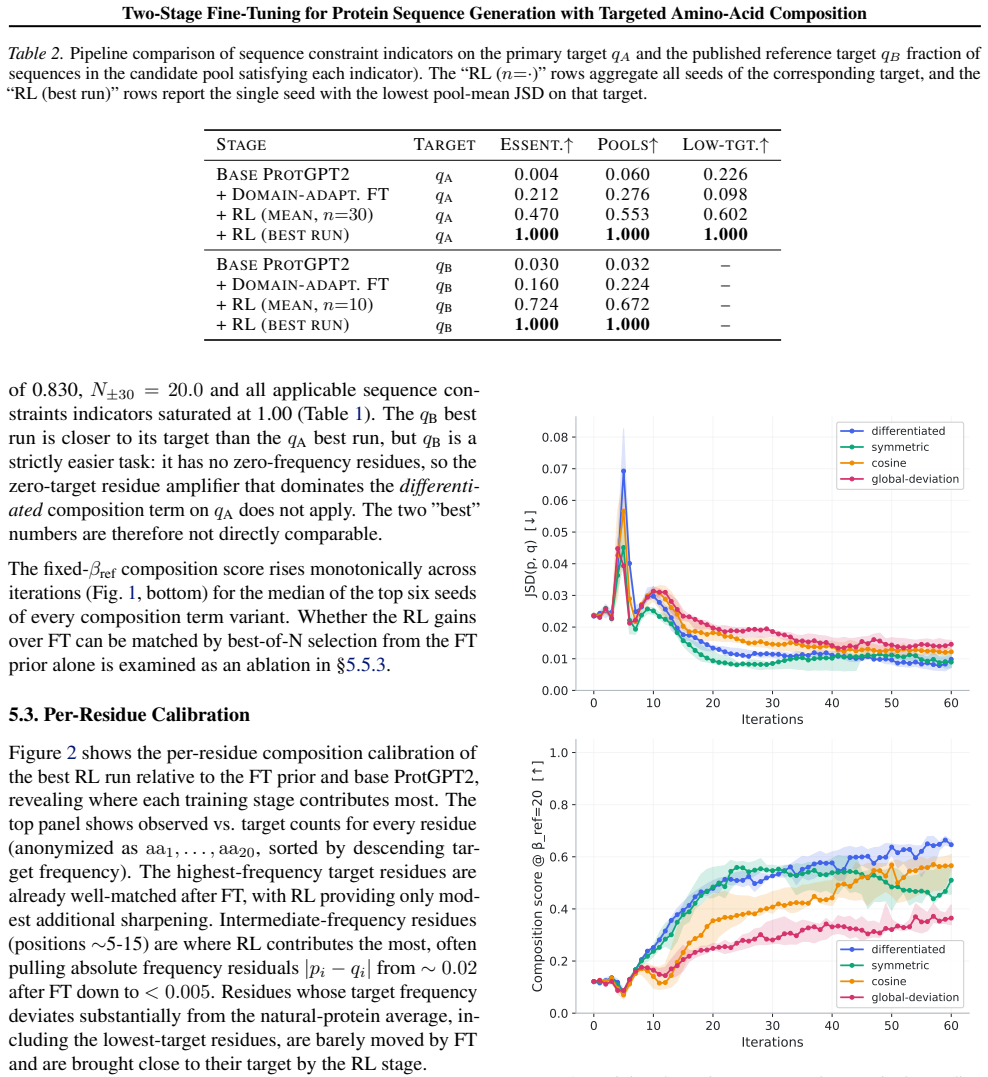
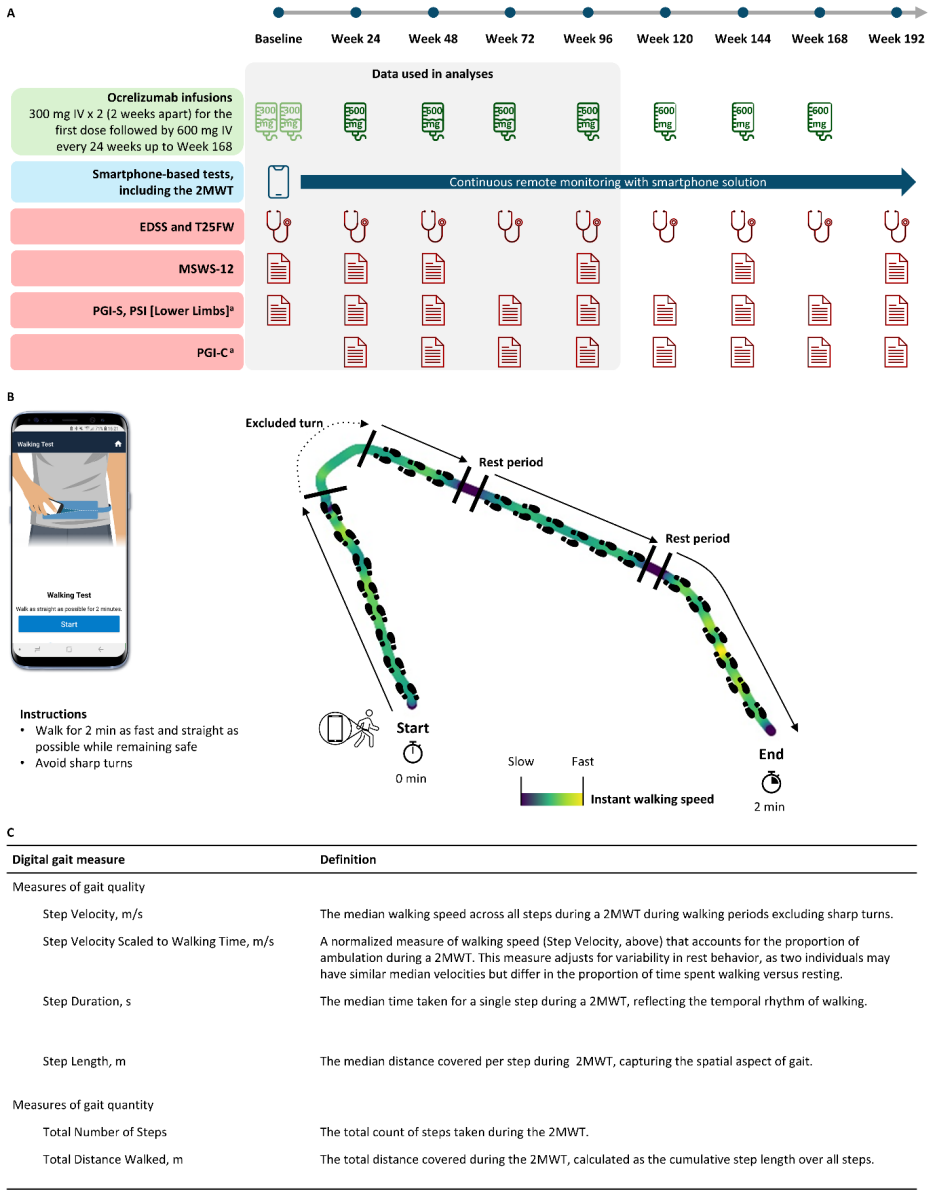
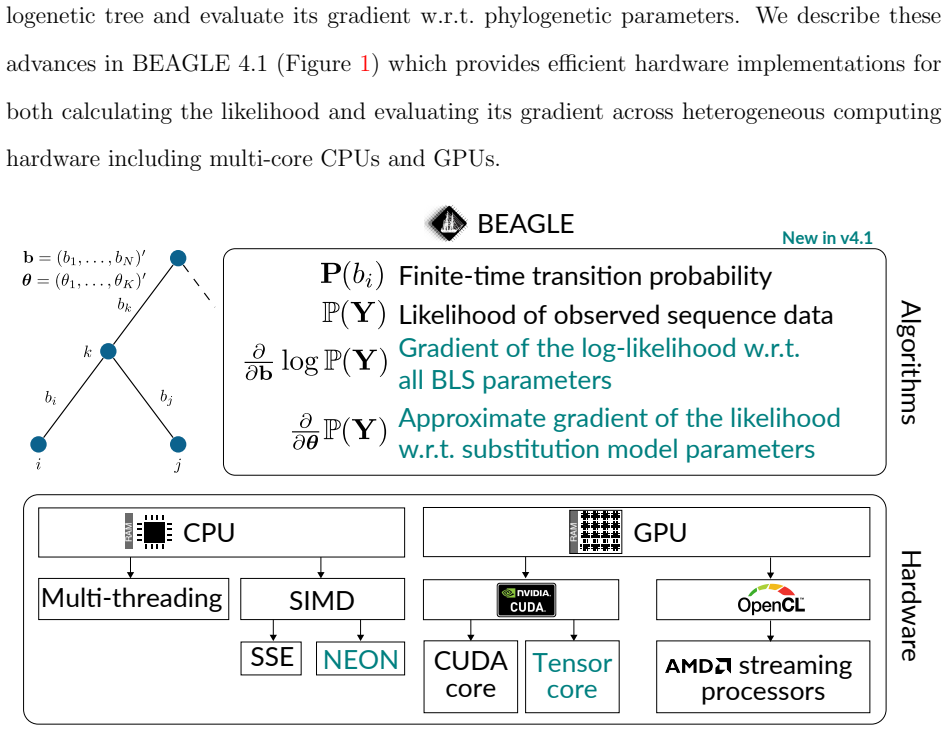
8
Lorentzian family is unique invariant under Riccati transport
Geometric Origin of Exact Mean-Field Reductions: M{\"o}bius Symmetry and the Lorentzian Ansatz
Reformulating dynamics on the circle shows the Cauchy law is the sole rotation-invariant measure, unifying exact mean-field reductions.
 full image
full image
abstract click to expand
Low-dimensional descriptions of large systems of coupled oscillators and spiking neurons rely heavily on the Lorentzian Ansatz. We show that its privileged role is geometric rather than heuristic: for the transport induced by Riccati dynamics, the Cauchy-Lorentz family indeed emerges as the unique connected two-dimensional family of continuous probability densities that is invariant under the induced projective transport. The key step of the demonstration is to reformulate the dynamics on the circle, where the problem reduces to the uniqueness of the rotation-invariant probability measure. Under stereographic projection, this yields the standard Cauchy law and, under the full projective action, the Lorentzian family. This result gives a unified geometric foundation for the Ott-Antonsen [Chaos 18, 037113 (2008)] and Montbri{\'o}-Paz{\'o}-Roxin [Phys. Rev. X 5, 021028 (2015)] reductions, explains the failure of Gaussian closures, and identifies the structural condition underlying exact two-parameter reductions.