DFT-assisted natural abundance 13C zero-field NMR via optical magnetometry
Pith reviewed 2026-05-07 14:17 UTC · model grok-4.3
The pith
Natural-abundance 13C zero-field NMR spectra are obtained from ordinary liquids using a compact commercial rubidium magnetometer and predicted accurately by vibrationally corrected density functional theory.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
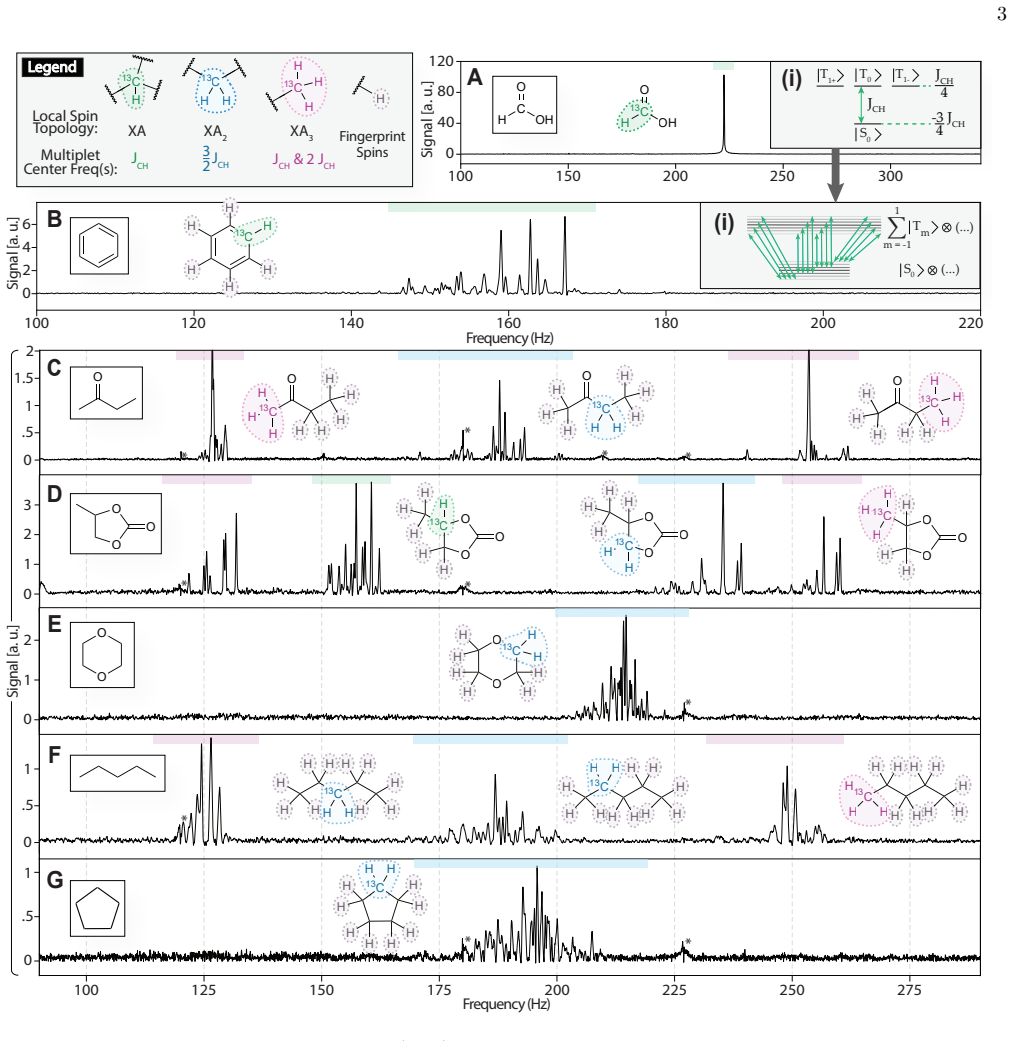
We demonstrate natural-abundance (1.1%) 13C ZF spectroscopy on off-the-shelf liquids using a compact commercial 87Rb magnetometer for the first time, without hyperpolarization or special sample preparation. Instrumental advances yield improved sensitivity, <250-mHz linewidths and >week-long stability, enabling isotopomer-resolved fingerprint spectra across a 13-molecule library, including the ability to discern rare (0.0121%) doubly 13C-labelled species. In parallel, we demonstrate vibrationally corrected density-functional theory (DFT) based prediction of ZF NMR spectra for chemically diverse molecules with few-hertz accuracy. Comparing experiment with these calculations renders residualdev
What carries the argument
Vibrationally corrected density-functional theory predictions of J-couplings combined with zero-field spectra detected by an 87Rb optical magnetometer.
Load-bearing premise
The assumption that vibrational corrections alone suffice for DFT to predict the J-couplings to few-hertz accuracy without molecule-specific adjustments.
What would settle it
A measurement on a new molecule where the experimental zero-field spectrum deviates by more than a few hertz from the vibrationally corrected DFT prediction after accounting for all known couplings.
Figures




read the original abstract
Zero-field (ZF) nuclear magnetic resonance (NMR) spectroscopy probes scalar J-couplings between nuclei while dispensing with large homogeneous magnetic fields, enabling low-cost and geometrically flexible detection, including through conductive enclosures. Despite these advantages, its broader use for chemical analysis has been limited by sensitivity and by the difficulty of predicting the dense spectral multiplets that arise at zero field. Here we demonstrate natural-abundance (1.1%) 13C ZF spectroscopy on off-the-shelf liquids using a compact commercial 87Rb magnetometer for the first time, without hyperpolarization or special sample preparation. Instrumental advances yield improved sensitivity, <250-mHz linewidths and >week-long stability, enabling isotopomer-resolved fingerprint spectra across a 13-molecule library, including the ability to discern rare (0.0121%) doubly 13C-labelled species. In parallel, we demonstrate vibrationally corrected density-functional theory (DFT) based prediction of ZF NMR spectra for chemically diverse molecules with few-hertz accuracy. Comparing experiment with these calculations renders residual deviations as chemically informative, reporting on hydrogen bonding, hydration and ion pairing at high ionic strength. Together, these results contribute towards DFT-assisted ZF NMR as a general platform for field-constraint-free molecular identification and for extracting transient solution-state structure from responsive J-coupling observables.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper demonstrates natural-abundance (1.1%) 13C zero-field NMR spectroscopy on standard liquid samples using a compact commercial 87Rb optical magnetometer, without hyperpolarization or special preparation. Instrumental improvements enable <250 mHz linewidths and week-long stability, yielding isotopomer-resolved spectra for a 13-molecule library. In parallel, vibrationally corrected DFT calculations are shown to predict the ZF spectra with few-Hz accuracy, allowing experimental residuals to be interpreted as signatures of hydrogen bonding, hydration, and ion pairing.
Significance. If the reported experimental sensitivity and DFT prediction accuracy hold, the work would establish a practical, low-cost platform for field-free molecular fingerprinting and for using J-couplings to probe transient solution structure. The combination of commercial hardware with parameter-free (post-vibrational-correction) theory is a notable strength that could broaden ZF NMR adoption in chemical analysis.
major comments (2)
- [DFT prediction and comparison section] The central claim of few-Hz DFT accuracy across chemically diverse molecules rests on the assertion that vibrational corrections alone suffice without molecule-specific fitting or functional tuning. However, no table or figure in the results section provides a complete side-by-side list of all experimental versus calculated J-couplings (with residuals and uncertainties) for the 13-molecule library, making it impossible to verify that the reported accuracy is achieved uniformly and without implicit parameter adjustment.
- [Experimental results and methods] The experimental sensitivity claims (natural-abundance 13C detection at <250 mHz linewidths with week-long stability on a commercial magnetometer) are load-bearing for the demonstration. The methods and results sections lack quantitative supporting data such as signal-to-noise ratios, acquisition parameters, and time-series stability metrics that would confirm these performance levels are routinely achieved for the reported spectra.
minor comments (2)
- [Figures] Figure captions and axis labels should explicitly state whether the displayed spectra are raw, processed, or simulated, and include the number of averages or total acquisition time.
- [Abstract and introduction] The abstract states 'few-hertz accuracy' but the main text should clarify the precise metric (RMS residual, maximum deviation, etc.) and the reference set of molecules used for validation.
Simulated Author's Rebuttal
We thank the referee for their constructive comments and positive overall assessment. We address each major point below and have revised the manuscript to improve verifiability of both the DFT predictions and the experimental performance metrics.
read point-by-point responses
-
Referee: [DFT prediction and comparison section] The central claim of few-Hz DFT accuracy across chemically diverse molecules rests on the assertion that vibrational corrections alone suffice without molecule-specific fitting or functional tuning. However, no table or figure in the results section provides a complete side-by-side list of all experimental versus calculated J-couplings (with residuals and uncertainties) for the 13-molecule library, making it impossible to verify that the reported accuracy is achieved uniformly and without implicit parameter adjustment.
Authors: We agree that a single consolidated table would facilitate independent verification. Although the per-molecule comparisons appear in the figures, the revised Supplementary Information now includes Table S2, which tabulates every experimental and vibrationally corrected DFT J-coupling for all 13 molecules together with residuals and experimental uncertainties (derived from observed linewidths). The vibrational corrections were obtained with a uniform computational protocol (B3LYP/6-311++G(d,p) with fixed scaling factors) applied identically to every species; no molecule-specific parameters or functional adjustments were introduced. This addition directly demonstrates that the few-hertz accuracy is achieved uniformly without implicit fitting. revision: yes
-
Referee: [Experimental results and methods] The experimental sensitivity claims (natural-abundance 13C detection at <250 mHz linewidths with week-long stability on a commercial magnetometer) are load-bearing for the demonstration. The methods and results sections lack quantitative supporting data such as signal-to-noise ratios, acquisition parameters, and time-series stability metrics that would confirm these performance levels are routinely achieved for the reported spectra.
Authors: We acknowledge that additional quantitative metrics strengthen the experimental claims. The revised Methods section now specifies acquisition parameters (typical averaging of 100–500 transients, magnetometer bandwidth, and 0.5 mL sample volumes), while the Results section reports representative signal-to-noise ratios. A new supplementary figure (Fig. S4) presents multi-day time-series data and Allan-deviation analysis confirming that linewidths remain below 250 mHz and that stability is maintained over at least seven days under routine operating conditions. These additions substantiate that the stated performance levels are routinely achieved for the library spectra. revision: yes
Circularity Check
No significant circularity; experimental spectra compared to independent DFT predictions
full rationale
The paper's central claims rest on direct experimental acquisition of natural-abundance 13C ZF spectra using a commercial Rb magnetometer and separate comparison to vibrationally corrected DFT calculations performed with standard methods. No equations or procedures are shown that define the predicted spectra in terms of the measured data, nor do any 'predictions' reduce to fitted parameters from the same dataset. Self-citations, if present for instrumental or computational details, are not load-bearing for the uniqueness or accuracy claims, as the DFT results remain falsifiable against external benchmarks and the experimental spectra are acquired independently. The derivation chain is therefore self-contained and externally validated.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard assumptions underlying density functional theory calculations of molecular electronic structure and vibrational corrections
Reference graph
Works this paper leans on
- [1]
-
[2]
R. R. Ernst, G. Bodenhausen, and A. Wokaun,Prin- ciples of Nuclear Magnetic Resonance in One and Two Dimensions(Oxford University PressOxford, 1990)
work page 1990
-
[3]
B. Shapira and L. Frydman, Journal of the American Chemical Society126, 7184 (2004)
work page 2004
-
[4]
P. C. Van Zijl, Journal of Magnetic Resonance (1969)75, 335 (1987)
work page 1969
-
[5]
J. W. Blanchard, M. P. Ledbetter, T. Theis, M. C. But- ler, D. Budker, and A. Pines, Journal of the American 10 Chemical Society135, 3607 (2013)
work page 2013
-
[6]
D. B. Zax, A. Bielecki, K. W. Zilm, A. Pines, and D. P. Weitekamp, The Journal of Chemical Physics83, 4877 (1985)
work page 1985
-
[7]
D. P. Weitekamp, A. Bielecki, D. Zax, K. Zilm, and A. Pines, Physical Review Letters50, 1807 (1983)
work page 1983
-
[8]
M. C. D. Tayler, T. Theis, T. F. Sjolander, J. W. Blan- chard, A. Kentner, S. Pustelny, A. Pines, and D. Budker, Review of Scientific Instruments88, 091101 (2017)
work page 2017
-
[9]
M. C. Tayler and D. Sakellariou, Journal of Magnetic Resonance277, 143 (2017)
work page 2017
- [10]
-
[11]
B. Andrews, M. Lai, Z. Wang, N. Kato, M. C. D. Tayler, E. Druga, and A. Ajoy, PNAS Nexus4, pgaf187 (2025)
work page 2025
-
[12]
D. B. Burueva, J. Eills, J. W. Blanchard, A. Garcon, R. Picazo-Frutos, K. V. Kovtunov, I. V. Koptyug, and D. Budker, Angewandte Chemie International Edition 59, 17026 (2020)
work page 2020
-
[13]
S. Bodenstedt, M. W. Mitchell, and M. C. D. Tayler, Nature Communications12, 4041 (2021)
work page 2021
- [14]
-
[15]
J. W. Blanchard and D. Budker, ineMagRes, edited by R. K. Harris and R. L. Wasylishen (John Wiley & Sons, Ltd, Chichester, UK, 2016) pp. 1395–1410
work page 2016
-
[16]
J. W. Blanchard, T. Wu, J. Eills, Y. Hu, and D. Budker, Journal of Magnetic Resonance314, 106723 (2020)
work page 2020
-
[17]
T. Wu, J. W. Blanchard, G. P. Centers, N. L. Figueroa, A. Garcon, P. W. Graham, D. F. J. Kimball, S. Rajen- dran, Y. V. Stadnik, A. O. Sushkov, A. Wickenbrock, and D. Budker, Physical Review Letters122, 191302 (2019)
work page 2019
-
[18]
A directly observable, Zeeman-insensitive nuclear spin coherence in solution,
J. Eills, A. Singh, A.-M. Teimoori, I. Marco-Rius, M. W. Mitchell, and M. C. D. Tayler, “A directly observable, Zeeman-insensitive nuclear spin coherence in solution,” (2026), arXiv:2601.07614 [physics]
-
[19]
D. A. Barskiy, M. C. D. Tayler, I. Marco-Rius, J. Kurhanewicz, D. B. Vigneron, S. Cikrikci, A. Ay- dogdu, M. Reh, A. N. Pravdivtsev, J.-B. H¨ ovener, J. W. Blanchard, T. Wu, D. Budker, and A. Pines, Nature Communications10, 3002 (2019)
work page 2019
-
[20]
R. Picazo-Frutos, K. F. Sheberstov, J. W. Blanchard, E. Van Dyke, M. Reh, T. Sjoelander, A. Pines, D. Bud- ker, and D. A. Barskiy, Nature Communications15, 4487 (2024)
work page 2024
-
[21]
M. C. Butler, M. P. Ledbetter, T. Theis, J. W. Blan- chard, D. Budker, and A. Pines, The Journal of Chemi- cal Physics138, 184202 (2013)
work page 2013
-
[22]
I. Mandzhieva, F. Theiss, X. He, A. Ortmeier, A. Koirala, S. J. McBride, S. J. DeVience, M. S. Rosen, V. Blum, and T. Theis, Journal of Chemical Information and Mod- eling65, 7554 (2025)
work page 2025
-
[23]
T. Helgaker, M. Jaszu´ nski, and M. Pecul, Progress in Nuclear Magnetic Resonance Spectroscopy53, 249 (2008)
work page 2008
- [24]
-
[25]
M. C. Tayler and S. Bodenstedt, Journal of Magnetic Resonance362, 107665 (2024)
work page 2024
-
[26]
D. A. Barskiy, J. W. Blanchard, D. Budker, J. Eills, S. Pustelny, K. F. Sheberstov, M. C. Tayler, and A. H. Trabesinger, Progress in Nuclear Magnetic Resonance Spectroscopy148–149, 101558 (2025)
work page 2025
-
[27]
M. J. Hadad, W. Zhang, T. Turney, L. Sernau, X. Wang, R. J. Woods, A. Incandela, I. Surjancev, A. Wang, M.-K. Yoon, A. Coscia, C. Euell, R. Meredith, I. Carmichael, and A. S. Serianni, inNMR in Glycoscience and Gly- cotechnology, edited by K. Kato and T. Peters (The Royal Society of Chemistry, 2017) pp. 20–100
work page 2017
-
[28]
Sternhell, Quarterly Reviews, Chemical Society23, 236 (1969)
S. Sternhell, Quarterly Reviews, Chemical Society23, 236 (1969)
work page 1969
- [29]
-
[30]
A. Wilzewski, S. Afach, J. Blanchard, and D. Budker, Journal of Magnetic Resonance284, 66 (2017)
work page 2017
-
[31]
S. Bodenstedt, M. W. Mitchell, and M. C. D. Tayler, Optically Detected Nuclear Magnetic Resonance above and Far below Earth’s Magnetic Field: Spin Dynam- ics and Relaxation in Unconventional Regimes, Ph.D. thesis, Universitat Polit` ecnica de Catalunya (2024), 2117/406472
work page 2024
-
[32]
D. M. Doddrell, D. T. Pegg, and M. R. Bendall, Journal of Magnetic Resonance (1969)48, 323 (1982)
work page 1969
-
[33]
G. M. Hale and M. R. Querry, Applied Optics12, 555 (1973)
work page 1973
-
[34]
D. I. Hoult and R. E. Richards, Journal of Magnetic Res- onance (1969)24, 71 (1976)
work page 1969
- [35]
-
[36]
H. J. Hogben, M. Krzystyniak, G. T. P. Charnock, P. J. Hore, and I. Kuprov, Journal of Magnetic Resonance 208, 179 (2011)
work page 2011
-
[37]
T. A. Ruden, O. B. Lutnæs, T. Helgaker, and K. Ruud, Journal of Chemical Physics118, 9572 (2003)
work page 2003
- [38]
-
[39]
Y. Shimizu, J. Blanchard, S. Pustelny, G. Saielli, A. Bagno, M. Ledbetter, D. Budker, and A. Pines, Jour- nal of Magnetic Resonance250, 1 (2015)
work page 2015
-
[40]
M. A. Z. Chowdhury, T. E. Rice, and M. A. Oehlschlaeger, Applied Physics B127, 34 (2021)
work page 2021
-
[41]
M. McCarthy and K. L. K. Lee, The Journal of Physical Chemistry A124, 3002 (2020)
work page 2020
-
[42]
H. Horai, M. Arita, S. Kanaya, Y. Nihei, T. Ikeda, K. Suwa, Y. Ojima, K. Tanaka, S. Tanaka, K. Aoshima, Y. Oda, Y. Kakazu, M. Kusano, T. Tohge, F. Mat- suda, Y. Sawada, M. Y. Hirai, H. Nakanishi, K. Ikeda, N. Akimoto, T. Maoka, H. Takahashi, T. Ara, N. Sakurai, H. Suzuki, D. Shibata, S. Neumann, T. Iida, K. Tanaka, K. Funatsu, F. Matsuura, T. Soga, R. Tag...
work page 2010
-
[43]
Scheiner,Hydrogen bonding: a theoretical perspective, Vol
S. Scheiner,Hydrogen bonding: a theoretical perspective, Vol. 7 (Oxford University Press, 1997)
work page 1997
- [44]
-
[45]
T. Giovannini, F. Egidi, and C. Cappelli, Chemical So- ciety Reviews49, 5664 (2020)
work page 2020
-
[46]
H. M. Rahman, G. Hefter, and R. Buchner, Journal of Physical Chemistry B116, 314 (2012)
work page 2012
-
[47]
K. Leung and S. B. Rempe, Journal of the American Chemical Society126, 344 (2004)
work page 2004
-
[48]
W. W. Rudolph and G. Irmer, Journal of Solution Chem- istry51, 935 (2022)
work page 2022
-
[49]
K. Trapp, S. Kosasang, J. Ingenmey, D. Gomez Vazquez, M. Salanne, and M. Lukatskaya, ChemRxiv (2025), 11 https://doi.org/10.26434/chemrxiv-2025-7j1vd-v2
-
[50]
E. A. Meyer, R. K. Castellano, and F. Diederich, Ange- wandte Chemie International Edition42, 1210 (2003)
work page 2003
-
[51]
P. Zhou, J. Huang, and F. Tian, Current medicinal chemistry19, 226 (2012)
work page 2012
-
[52]
T. Hong, Y. Wang, Z. Shao, Q. Li, M. Jiang, and X. Peng, Magnetic Resonance Letters5, 200170 (2025)
work page 2025
-
[53]
P.-H. Chen, C. Gao, N. Alaniva, S. Bj¨ orgvinsd´ ottir, I. G. Pagonakis, M. A. Urban, A. D¨ app, R. Gunzenhauser, and A. B. Barnes, Journal of Magnetic Resonance357, 107588 (2023)
work page 2023
-
[54]
W. Lee, D. Park, J. Bascu˜ n´ an, and Y. Iwasa, Supercon- ductor Science and Technology35, 105007 (2022)
work page 2022
-
[55]
Neese, WIREs Computational Molecular Science15, e70019 (2025)
F. Neese, WIREs Computational Molecular Science15, e70019 (2025)
work page 2025
-
[56]
B. Helmich-Paris, B. de Souza, F. Neese, and R. Izs´ ak, Journal of Chemical Physics155, 104109 (2021)
work page 2021
-
[57]
G. L. Stoychev, A. A. Auer, and F. Neese, Journal of Chemical Theory and Computation13, 554 (2017)
work page 2017
-
[58]
de Souza, Angewandte Chemie International Edition 64, e202500393 (2025)
B. de Souza, Angewandte Chemie International Edition 64, e202500393 (2025)
work page 2025
-
[59]
C. Bannwarth, S. Ehlert, and S. Grimme, Journal of Chemical Theory and Computation15, 1652 (2019)
work page 2019
- [60]
-
[61]
T. H. Dunning Jr, Journal of Chemical Physics90, 1007 (1989)
work page 1989
-
[62]
R. A. Kendall, T. H. Dunning Jr, and R. J. Harrison, Journal of Chemical Physics96, 6796 (1992)
work page 1992
-
[63]
D. E. Woon and T. H. Dunning Jr, Journal of Chemical Physics103, 4572 (1995)
work page 1995
-
[64]
Jensen, Journal of Chemical Theory and Computation 2, 1360 (2006)
F. Jensen, Journal of Chemical Theory and Computation 2, 1360 (2006)
work page 2006
-
[65]
P. R. Franke, J. F. Stanton, and G. E. Douberly, Journal of Physical Chemistry A125, 1301 (2021)
work page 2021
-
[66]
F. Pavoˇ sevi´ c, F. Neese, and E. F. Valeev, Journal of chemical physics141, 054106 (2014)
work page 2014
-
[67]
K. A. Peterson, T. B. Adler, and H.-J. Werner, Journal of Chemical Physics128, 084102 (2008)
work page 2008
- [68]
-
[69]
Jensen, Journal of Physical Chemistry A111, 11198 (2007)
F. Jensen, Journal of Physical Chemistry A111, 11198 (2007)
work page 2007
-
[70]
Jensen, Journal of Chemical Physics136, 114107 (2012)
F. Jensen, Journal of Chemical Physics136, 114107 (2012)
work page 2012
-
[71]
P. R. Horn, Y. Mao, and M. Head-Gordon, Physical Chemistry Chemical Physics18, 23067 (2016)
work page 2016
-
[72]
E. Epifanovsky, A. T. Gilbert, X. Feng, J. Lee, Y. Mao, N. Mardirossian, P. Pokhilko, A. F. White, M. P. Coons, A. L. Dempwolff,et al., Journal of Chemical Physics155, 084801 (2021)
work page 2021
-
[73]
J.-D. Chai and M. Head-Gordon, Journal of Chemical Physics128, 084106 (2008)
work page 2008
-
[74]
J.-D. Chai and M. Head-Gordon, Physical Chemistry Chemical Physics10, 6615 (2008)
work page 2008
- [75]
-
[76]
F. Weigend and R. Ahlrichs, Physical Chemistry Chem- ical Physics7, 3297 (2005)
work page 2005
-
[77]
D. Rappoport and F. Furche, Journal of Chemical Physics133, 134105 (2010)
work page 2010
-
[78]
Y. Mao,Advances in Density Functional Theory Calcula- tions and Their Analysis(University of California, Berke- ley, 2017)
work page 2017
-
[79]
E. F. Valeev, Chemical Physics Letters395, 190 (2004)
work page 2004
-
[80]
F. Weigend, A. K¨ ohn, and C. H¨ attig, Journal of Chem- ical Physics116, 3175 (2002)
work page 2002
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.