Implementation of the hybrid exchange-correlation functionals in the SIESTA code
Pith reviewed 2026-05-07 15:45 UTC · model grok-4.3
The pith
Hybrid functionals are now implemented in SIESTA for large systems by fitting numerical atomic orbitals to Gaussians for exact exchange.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By representing numerical atomic orbitals as linear combinations of Gaussian-type orbitals, four-center electron-repulsion integrals can be computed analytically with LIBINT, allowing hybrid functionals to be evaluated efficiently inside SIESTA's sparse real-space infrastructure with controlled screening and scalable parallelism.
What carries the argument
Gaussian fit of numerical atomic orbitals enabling analytical four-center ERIs via LIBINT.
If this is right
- HSE06 calculations become practical for systems containing hundreds to thousands of atoms.
- Structural relaxations and molecular-dynamics runs can use hybrid functionals with analytic forces.
- Users can trade accuracy against cost by adjusting the number of Gaussians and the integral-screening thresholds.
- Results for semiconductors and 2D materials lie close to G0W0 and experiment.
Where Pith is reading between the lines
- The same fitting strategy could be reused to add other exact-exchange-based methods to NAO-based codes.
- Parallel scaling data imply that even larger supercells or defect supercells are now within reach.
- The reported trade-off curves can serve as a practical guide for choosing parameters on new materials.
Load-bearing premise
The Gaussian expansion reproduces the numerical orbitals accurately enough that the resulting exact-exchange contributions do not change band gaps or forces beyond acceptable numerical tolerances.
What would settle it
Direct comparison on a small periodic system where the hybrid band gap or force differs by more than a few percent from the same quantity computed with an all-electron code that uses exact numerical orbitals.
Figures
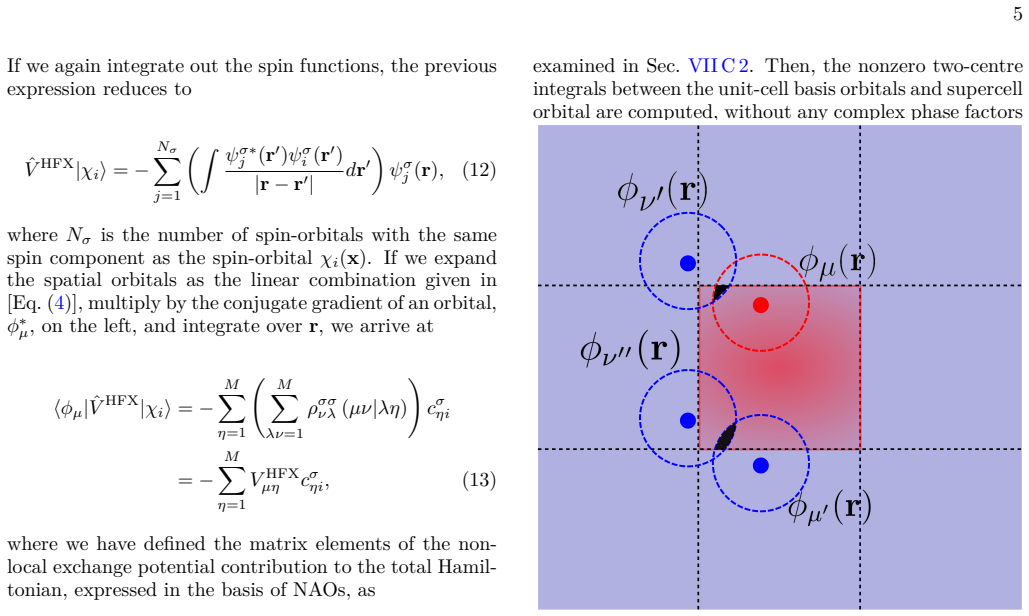


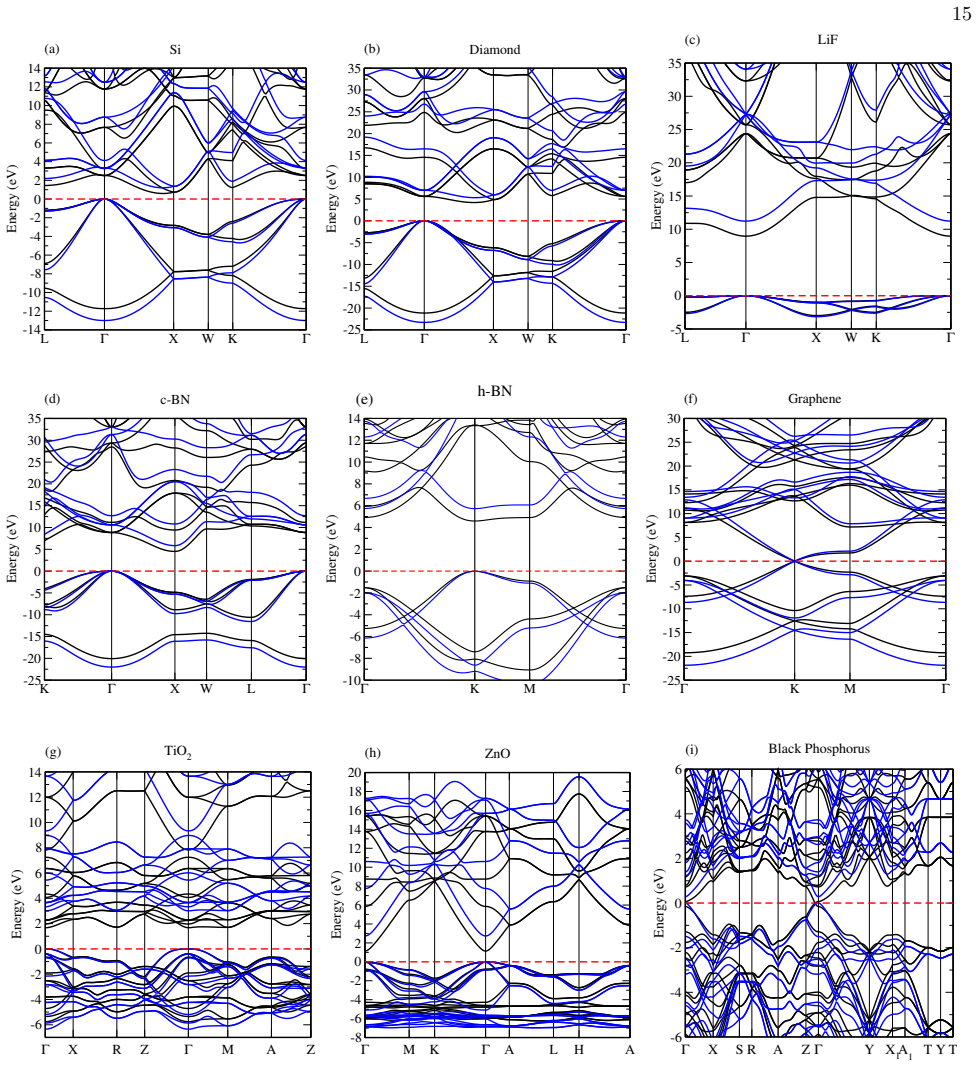

read the original abstract
We present an efficient and accurate implementation of hybrid exchange-correlation (XC) functionals in the SIESTA code, enabling large-scale simulations based on Hartree-Fock-type exact exchange combined with strictly localized numerical atomic orbitals (NAOs). Our approach exploits a fitted representation of the NAOs in terms of Gaussian-type orbitals (GTOs), which allows for the analytical evaluation of four-center electron repulsion integrals (ERIs) via the LIBINT library. This framework is seamlessly integrated with SIESTA's real-space grid and sparse-matrix infrastructure, and is combined with multiple screening techniques to control the computational complexity. We also introduce a fully analytical formulation of hybrid-functional forces and a dynamic parallel distribution scheme that ensures excellent scalability. We validate our implementation through benchmark calculations on a broad set of systems (including semiconductors, insulators, and two-dimensional materials) and demonstrate that the HSE06 functional significantly improves the prediction of band gaps compared to PBE, in close agreement with G0W0 and experimental data. We analyze in detail the trade-offs between accuracy and computational efficiency as a function of the number of Gaussians, basis set range, and integral screening thresholds. Our results confirm that hybrid functional calculations in SIESTA are now feasible for large extended systems, making accurate first-principles predictions of electronic and structural properties accessible at scale.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper describes an implementation of hybrid XC functionals (e.g., HSE06) in the SIESTA code. NAOs are fitted to GTOs to enable analytic four-center ERIs via LIBINT; this is combined with SIESTA's real-space grid, sparse matrices, integral screening, a fully analytic force formulation, and dynamic parallel distribution. Benchmarks on semiconductors, insulators, and 2D materials are reported to show that HSE06 band gaps improve over PBE and agree with G0W0 and experiment; trade-offs with number of Gaussians, basis range, and screening thresholds are analyzed.
Significance. If the NAO-to-GTO fit and screening preserve the accuracy of the exact-exchange contribution, the work would make hybrid-functional calculations routinely feasible for large periodic systems in a widely used localized-basis code, addressing a practical need in materials modeling where semilocal functionals often fail for band gaps and defect levels.
major comments (2)
- [§3] §3 (Implementation of the NAO-GTO fit and ERI evaluation): the central accuracy claim rests on the fitted GTO representation reproducing the exact-exchange matrix elements of the original NAOs to sufficient precision, yet no explicit error metric (e.g., maximum deviation in exchange energy or matrix elements relative to an unfitted NAO reference) is supplied as a function of the number of Gaussians; without this, agreement with external G0W0/experimental data could mask compensating errors.
- [§4] §4 (Benchmark calculations): while HSE06 results are stated to agree with G0W0 and experiment, the manuscript provides no side-by-side comparison against an independent hybrid-functional implementation (plane-wave or other NAO code) on the same periodic cells; such a cross-code validation would be required to confirm that fit-induced and screening-induced errors remain below the reported band-gap improvements.
minor comments (3)
- [§3.3] The description of the dynamic parallel distribution scheme would benefit from a short pseudocode or timing table showing load balance across nodes for a representative large system.
- [Figures 4-6] Figure captions for the basis-range and screening-threshold convergence plots should explicitly state the system, functional, and property (band gap or total energy) being plotted.
- [§3.2] A brief statement on the memory scaling of the LIBINT ERI storage relative to the sparse-matrix infrastructure would clarify the practical limits for very large cells.
Simulated Author's Rebuttal
We are grateful to the referee for the thorough review and constructive feedback on our manuscript. We have carefully considered the major comments and will revise the manuscript accordingly to address the concerns regarding the accuracy of the NAO-GTO fit and the need for cross-validation.
read point-by-point responses
-
Referee: [§3] §3 (Implementation of the NAO-GTO fit and ERI evaluation): the central accuracy claim rests on the fitted GTO representation reproducing the exact-exchange matrix elements of the original NAOs to sufficient precision, yet no explicit error metric (e.g., maximum deviation in exchange energy or matrix elements relative to an unfitted NAO reference) is supplied as a function of the number of Gaussians; without this, agreement with external G0W0/experimental data could mask compensating errors.
Authors: We thank the referee for highlighting this important point. While our manuscript analyzes the trade-offs between accuracy and efficiency as a function of the number of Gaussians (including effects on band gaps), we acknowledge that we did not provide a direct, explicit error metric for the reproduction of exact-exchange matrix elements or energies by the fitted GTOs compared to the original NAOs. To address this, in the revised manuscript we will include a dedicated analysis, such as a table or plot, showing the maximum deviation in the exchange energy and selected matrix elements as a function of the number of Gaussians for benchmark systems. This will demonstrate that the fitting errors are well-controlled and do not compromise the reported improvements. revision: yes
-
Referee: [§4] §4 (Benchmark calculations): while HSE06 results are stated to agree with G0W0 and experiment, the manuscript provides no side-by-side comparison against an independent hybrid-functional implementation (plane-wave or other NAO code) on the same periodic cells; such a cross-code validation would be required to confirm that fit-induced and screening-induced errors remain below the reported band-gap improvements.
Authors: We agree that direct cross-code validation would provide stronger evidence that the implementation errors are negligible compared to the functional improvements. Although our results show good agreement with G0W0 and experimental band gaps, this does not explicitly rule out compensating errors from the fit or screening. In the revision, we will add comparisons with results from established hybrid-functional implementations (such as VASP or other codes) for a selection of the smaller periodic systems studied, using identical unit cells and k-point sampling where possible. This will help confirm the reliability of our approach. revision: yes
Circularity Check
No circularity: implementation validated against external references
full rationale
The paper presents a software implementation of hybrid XC functionals in SIESTA via NAO-to-GTO fitting for analytic ERIs with LIBINT, combined with screening and parallelization. Its central feasibility claim for large systems rests on benchmark results for band gaps and forces that are compared directly to independent G0W0 calculations and experimental values, not to any quantity derived from the paper's own fitted parameters or prior self-citations. No derivation step reduces by construction to a fitted input renamed as a prediction, nor does any uniqueness theorem or ansatz depend on self-referential citations. The Gaussian-fit accuracy is treated as an engineering assumption whose quality is assessed externally rather than enforced internally.
Axiom & Free-Parameter Ledger
free parameters (2)
- number of Gaussians per NAO
- integral screening thresholds
axioms (1)
- domain assumption A Gaussian expansion of the numerical atomic orbitals permits sufficiently accurate analytical evaluation of the four-center electron-repulsion integrals required for exact exchange.
Reference graph
Works this paper leans on
-
[1]
within the PBE flavor, and available in the Pseudo- Dojo periodic table [ 129, 130]. Although these pseu- dopotentials are not strictly consistent with the hy- brid exchange–correlation functional, previous system- atic studies have shown that the resulting errors in struc- tural and electronic properties are typically small (of the order of 1%) and well ...
-
[2]
Number of Gaussians The number of Gaussians used to expand the radial components of the NAOs significantly impacts the effi- ciency of hybrid functional simulations, as illustrated in Table-II. A larger NG reduces the goal function that mea- sures the difference between the sum of Gaussians and the original radial part of the native NAOs [metric ˜G, define...
work page 2004
-
[3]
Range of the radial parts of the atomic orbitals The spatial extent of the NAOs plays a crucial role in determining the sparsity of the Hamiltonian matrix and, by extension, the computational efficiency of hybrid functional calculations. Table- IV explores the effect of varying the cutoff radius rc used to truncate the orbitals, controlled via the ϵGauss p...
work page 2046
-
[4]
Integral screening thresholds To assess the impact of integral screening on the accu- racy and efficiency of hybrid functional calculations, we perform a systematic benchmark by varying the screen- ing thresholds discussed in Sec. IV. In particular, we set the Schwarz pair-list threshold ϵpair−list and the density screening threshold ϵSchwartz [cf. Eqs. ( ...
-
[5]
P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136, B864 (1964)
work page 1964
-
[6]
R. M. Martin, Electronic Structure: Basic Theory and Practical Methods (Cambridge University Press, 2004)
work page 2004
-
[7]
J. Kohanoff, Electronic Structure Calculations for Solids and Molecules: Theory and Computational Methods (Cambridge University Press, 2006)
work page 2006
-
[8]
R. G. Parr and W. Yang, Density F unctional T heory of Atoms and M olecules (Oxford University Press, 1989)
work page 1989
-
[9]
W. Kohn and L. J. Sham, Self-consistent equations in- cluding exchange and correlation effects, Phys. Rev. 140, A1133 (1965)
work page 1965
-
[10]
D. M. Ceperley and B. J. Alder, Ground state of the electron gas by a stochastic method, Phys. Rev. Lett. 45, 566 (1980)
work page 1980
-
[11]
J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B 23, 5048 (1981)
work page 1981
-
[12]
J. P. Perdew and K. Burke, Comparison shopping for a gradient-corrected density functional, Int. J. Quant. Chem. 57, 309 (1996)
work page 1996
-
[13]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865 (1996)
work page 1996
-
[14]
J. P. Perdew, K. Burke, and M. Ernzerhof, Erratum: Generalized gradient approximation made simple [phys. rev. lett. 77, 3865 (1996)], Phys. Rev. Lett. 78, 1396 (1997)
work page 1996
-
[15]
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vy- drov, G. E. Scuseria, L. A. Constantin, X. Zhou, and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett. 100, 136406 (2008)
work page 2008
- [16]
-
[17]
J. P. Perdew and K. Schmidt, Jacob’s ladder of density functional approximations for the exchange-correlation energy, AIP Conf. Proc. 577, 1 (2001)
work page 2001
-
[18]
A. J. Cohen, P. Mori-Sánchez, and W. Yang, Insights into current limitations of density functional theory, Sci- ence 321, 792 (2008)
work page 2008
-
[19]
J. P. Perdew and M. Levy, Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities, Phys. Rev. Lett. 51, 1884 (1983)
work page 1983
-
[20]
L. J. Sham and M. Schlüter, Density-functional theory of the energy gap, Phys. Rev. Lett. 51, 1888 (1983)
work page 1983
-
[21]
P. Mori-Sánchez, A. J. Cohen, and W. Yang, Local- ization and delocalization errors in density functional theory and implications for band-gap prediction, Phys. Rev. Lett. 100, 146401 (2008)
work page 2008
-
[22]
V. Anisimov, Strong Coulomb Correlations in Electronic Structure Calculations , Advances in Condensed Matter 23 Science (Taylor & Francis, 2000)
work page 2000
-
[23]
A. D. Becke, A new mixing of Hartree-Fock and lo- cal density‐functional theories, J. Chem. Phys. 98, 1372 (1993)
work page 1993
-
[24]
P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch, Ab initio calculation of vibrational ab- sorption and circular dichroism spectra using density functional force fields, J. Phys. Chem. 98, 11623 (1994)
work page 1994
-
[25]
C. Adamo and V. Barone, Toward reliable density func- tional methods without adjustable parameters: The PBE0 model, J. Chem. Phys. 110, 6158 (1999)
work page 1999
-
[26]
J. Heyd, G. E. Scuseria, and M. Ernzerhof, Hybrid func- tionals based on a screened Coulomb potential, J. Chem. Phys. 118, 8207 (2003)
work page 2003
-
[27]
Hybrid functionals based on a screened Coulomb po- tential
J. Heyd, G. E. Scuseria, and M. Ernzerhof, Erratum: “Hybrid functionals based on a screened Coulomb po- tential” [ J C hem. Phys. 118, 8207 (2003)], J. Chem. Phys. 124, 219906 (2006)
work page 2003
-
[28]
A. V. Krukau, O. A. Vydrov, A. F. Izmaylov, and G. E. Scuseria, Influence of the exchange screening parameter on the performance of screened hybrid functionals, J. Chem. Phys. 125, 224106 (2006)
work page 2006
-
[29]
L. Schimka, J. Harl, and G. Kresse, Improved hybrid functional for solids: The HSEsol functional, J. Chem. Phys. 134, 024116 (2011)
work page 2011
-
[30]
B. G. Janesko, T. M. Henderson, and G. E. Scuse- ria, Screened hybrid density functionals for solid-state chemistry and physics, Phys. Chem. Chem. Phys. 11, 443 (2009)
work page 2009
-
[31]
A. J. Garza and G. E. Scuseria, Predicting band gaps with hybrid density functionals, J. Phys. Chem. Lett. 7, 4165 (2016)
work page 2016
-
[32]
P. Borlido, T. Aull, A. W. Huran, F. Tran, M. A. L. Marques, and S. Botti, Large-scale benchmark of exchange–correlation functionals for the determination of electronic band gaps of solids, J. Chem. Theory Com- put. 15, 5069 (2019)
work page 2019
-
[33]
J. Yang, S. Falletta, and A. Pasquarello, Range- separated hybrid functionals for accurate prediction of band gaps of extended systems, npj Comput. Mater. 9, 108 (2023)
work page 2023
- [34]
-
[35]
screened hybrid density functionals applied to solids
J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber, and J. G. Ángyán, Erratum: “screened hybrid density functionals applied to solids” [j. chem. phys. 124, 154709 (2006)], The Journal of Chemical Physics 125, 249901 (2006)
work page 2006
-
[36]
S. Lany and A. Zunger, Polaronic hole localization and multiple hole binding of acceptors in oxide wide-gap semiconductors, Phys. Rev. B 80, 085202 (2009)
work page 2009
-
[37]
W. Chen and A. Pasquarello, Correspondence of de- fect energy levels in hybrid density functional theory and many-body perturbation theory, Phys. Rev. B 88, 115104 (2013)
work page 2013
-
[38]
W. Chen and A. Pasquarello, Accuracy of GW for cal- culating defect energy levels in solids, Phys. Rev. B 96, 020101 (2017)
work page 2017
-
[39]
H. Guo, X. Zhang, and G. Lu, Moiré excitons in defec- tive van der Waals heterostructures, Proc. Natl. Acad. Sci. U.S.A. 118, e2105468118 (2021)
work page 2021
-
[40]
W. Chen, S. M. Griffin, G.-M. Rignanese, and G. Hau- tier, Nonunique fraction of Fock exchange for defects in two-dimensional materials, Phys. Rev. B 106, L161107 (2022)
work page 2022
-
[41]
S. Ke, S. E. Gant, L. Kronik, and J. B. Neaton, Accurate point defect energy levels from non-empirical screened range-separated hybrid functionals: The case of native vacancies in ZnO, Phys. Rev. Mater. 9, 053806 (2025)
work page 2025
-
[42]
F. Sagredo, M. Camarasa-Gómez, F. Ricci, A. Cham- pagne, L. Kronik, and J. B. Neaton, The reliability of hybrid functionals for accurate fundamental and opti- cal gap prediction of bulk solids and surfaces, J. Chem. Theory Comput. 21, 5009 (2025)
work page 2025
-
[43]
X. Gonze, F. Jollet, F. Abreu Araujo, D. Adams, B. Amadon, T. Applencourt, C. Audouze, J.-M. Beuken, J. Bieder, A. Bokhanchuk, E. Bousquet, F. Bruneval, D. Caliste, M. Côté, F. Dahm, F. Da Pieve, M. Delaveau, M. Di Gennaro, B. Dorado, C. Es- pejo, G. Geneste, L. Genovese, A. Gerossier, M. Gi- antomassi, Y. Gillet, D. Hamann, L. He, G. Jomard, J. Laflamme ...
work page 2016
-
[44]
Lin, Adaptively compressed exchange operator, J
L. Lin, Adaptively compressed exchange operator, J. Chem. Theory Comput. 12, 2242 (2016)
work page 2016
-
[45]
I. Duchemin and F. Gygi, A scalable and accurate al- gorithm for the computation of Hartree–Fock exchange, Comput. Phys. Commun. 181, 855 (2010)
work page 2010
-
[46]
R. A. DiStasio, B. Santra, Z. Li, X. Wu, and R. Car, The individual and collective effects of exact exchange and dispersion interactions on the ab initio structure of liquid water, J. Chem. Phys. 141, 084502 (2014)
work page 2014
-
[47]
T. A. Barnes, T. Kurth, P. Carrier, N. Wichmann, D. Prendergast, P. R. Kent, and J. Deslippe, Improved treatment of exact exchange in Quantum espresso, Comput. Phys. Commun. 214, 52 (2017)
work page 2017
-
[48]
X. Wu, A. Selloni, and R. Car, Order- N implementation of exact exchange in extended insulating systems, Phys. Rev. B 79, 085102 (2009)
work page 2009
-
[49]
A. Natan, Fock-exchange for periodic structures in the real-space formalism and the KLI approximation, Phys. Chem. Chem. Phys. 17, 31510 (2015)
work page 2015
-
[50]
N. M. Boffi, M. Jain, and A. Natan, Efficient Computa- tion of the Hartree–Fock Exchange in Real-Space with Projection Operators, J. Chem. Theory Comput. 12, 3614 (2016)
work page 2016
-
[51]
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scal- mani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnen- berg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Ki- tao, H. N...
work page 2009
- [52]
-
[53]
J. VandeVondele, M. Krack, F. Mohamed, M. Par- rinello, T. Chassaing, and J. Hutter, Quickstep: Fast and accurate density functional calculations using a mixed gaussian and plane waves approach, Comput. Phys. Commun. 167, 103 (2005)
work page 2005
-
[54]
I. J. Bush, S. Tomic, B. G. Searle, G. Mallia, C. L. Bailey, B. Montanari, L. Bernasconi, J. M. Carr, and N. M. Harrison, Parallel implementation of the ab initio crystal program: electronic structure calculations for periodic systems, Proc. R. Soc. A 467, 2112 (2011)
work page 2011
-
[55]
G. t. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Sni- jders, and T. Ziegler, Chemistry with ADF, J. Comput. Chem. 22, 931 (2001)
work page 2001
-
[56]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler, Ab initio molec- ular simulations with numeric atom-centered orbitals, Comput. Phys. Commun. 180, 2175 (2009)
work page 2009
-
[57]
V. Havu, V. Blum, P. Havu, and M. Scheffler, Efficient integration for all-electron electronic structure calcula- tion using numeric basis functions, J. Comput. Phys. 228, 8367 (2009)
work page 2009
-
[58]
X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter, and M. Schef- fler, Resolution-of-identity approach to Hartree–Fock, hybrid density functionals, RPA, MP2 and GW with numeric atom-centered orbital basis functions, New J. Phys. 14, 053020 (2012)
work page 2012
-
[59]
X. Qin, H. Shang, L. Xu, W. Hu, J. Yang, S. Li, and Y. Zhang, The static parallel distribution algorithms for hybrid density-functional calculations in honpas package, Int. J. High Perform. Comput. Appl. 34, 159 (2019)
work page 2019
- [60]
-
[61]
E. Schwegler and M. Challacombe, Linear scaling com- putation of the Hartree–Fock exchange matrix, J. Chem. Phys. 105, 2726 (1996)
work page 1996
-
[62]
J. C. Burant, G. E. Scuseria, and M. J. Frisch, A linear scaling method for Hartree–Fock exchange calculations of large molecules, J. Chem. Phys. 105, 8969 (1996)
work page 1996
-
[63]
E. Schwegler, M. Challacombe, and M. Head-Gordon, Linear scaling computation of the Fock matrix. II. rigor- ous bounds on exchange integrals and incremental Fock build, J. Chem. Phys. 106, 9708 (1997)
work page 1997
-
[64]
C. Ochsenfeld, C. A. White, and M. Head-Gordon, Lin- ear and sublinear scaling formation of Hartree–Fock- type exchange matrices, J. Chem. Phys. 109, 1663 (1998)
work page 1998
-
[65]
R. Polly, H.-J. Werner, F. R. Manby, and P. J. Knowles, Fast Hartree–Fock theory using local density fitting ap- proximations, Mol. Phys. 102, 2311 (2004)
work page 2004
-
[66]
A. Sodt and M. Head-Gordon, Hartree-Fock exchange computed using the atomic resolution of the identity approximation, J. Chem. Phys. 128, 104106 (2008)
work page 2008
- [67]
-
[68]
X. Liu, A. Patel, and E. Chow, A new scalable parallel algorithm for Fock matrix construction, in 2014 IEEE 28th International Parallel and Distributed Processing Symposium (IEEE, 2014)
work page 2014
-
[69]
E. Chow, X. Liu, M. Smelyanskiy, and J. R. Ham- mond, Parallel scalability of Hartree–Fock calculations, J. Chem. Phys. 142, 104103 (2015)
work page 2015
-
[70]
See: http://gaussian.com
- [71]
- [72]
-
[73]
T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Ry- bkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, et al. , cp2k: An electronic structure and molecular dynamics software package- quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys. 152, 194103 (2020)
work page 2020
-
[74]
M. Iannuzzi, J. Wilhelm, F. Stein, A. Bussy, H. El- gabarty, D. Golze, A. Hehn, M. Graml, S. Marek, B. S. Gökmen, C. Schran, H. Forbert, R. Z. Khaliullin, A. Kozhevnikov, M. Taillefumier, R. Meli, V. Rybkin, M. Brehm, R. Schade, O. Schütt, J. V. Pototschnig, H. Mirhosseini, A. Knüpfer, D. Marx, M. Krack, J. Hut- ter, and T. D. Kühne, The cp2k program pack...
-
[75]
See: https://www.cp2k.org
-
[76]
I. J. Bush, S. Tomić, B. G. Searle, G. Mallia, C. L. Bailey, B. Montanari, L. Bernasconi, J. M. Carr, and N. M. Harrison, Parallel implementation of the ab initio crystal program: Electronic structure calculations for periodic systems, Proc. R. Soc. A 467, 2112 (2011)
work page 2011
-
[77]
R. Dovesi, F. Pascale, B. Civalleri, K. Doll, N. M. Har- rison, I. Bush, P. D’Arco, Y. Noël, M. Rérat, P. Car- bonnière, M. Causà, S. Salustro, V. Lacivita, B. Kirt- man, A. M. Ferrari, F. S. Gentile, J. Baima, M. Ferrero, R. Demichelis, and M. De La Pierre, The crystal code, 1976–2020 and beyond, a long story, J. Chem. Phys. 152, 204111 (2020)
work page 1976
-
[78]
A. Erba, J. K. Desmarais, S. Casassa, B. Civalleri, L. Donà, I. J. Bush, B. Searle, L. Maschio, L. Edith- Daga, A. Cossard, et al. , crystal23: A program for computational solid state physics and chemistry, J. Chem. Theory Comput. 19, 6891 (2022)
work page 2022
-
[79]
See: https://www.crystal.unito.it/index.php
-
[80]
J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Jun- quera, P. Ordejón, and D. Sánchez-Portal, The siesta method for ab initio order-N materials simulation, J. Phys.: Condens. Matter 14, 2745 (2002)
work page 2002
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.