Radical-Fragment Many-Body Expansion for Linear Alkane Quantum Chemistry
Pith reviewed 2026-07-01 03:26 UTC · model grok-4.3
The pith
A two-body radical-fragment expansion reconstructs linear alkane energies from four unique calculations, cutting qubit needs by over 12 times for C26H54.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Linear alkane total energies can be assembled from two-body interactions of homolytic radical fragments (CH3 and CH2) computed in isolation with ROHF, using an assembly formula that requires only four unique fragment calculations independent of chain length.
What carries the argument
The two-body MBE2 assembly formula applied to energies of homolytic C-C cleavage radical fragments treated without capping atoms or embedding.
If this is right
- Total energies for linear alkanes of any length are obtained from four unique fragment calculations.
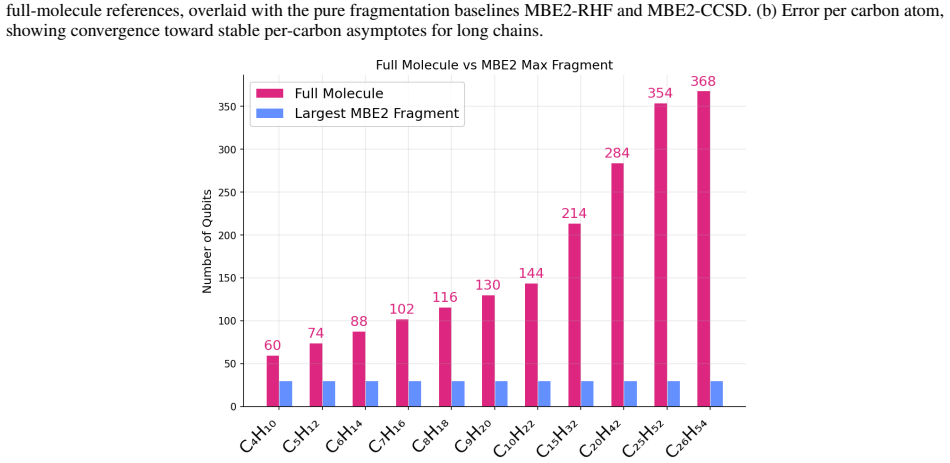
- Maximum qubit requirement drops with fragment size rather than full molecule size, yielding a 12.3x reduction for C26H54.
- MBE2-VQE and MBE2-SQD executed on IBM hardware closely track their classical MBE2 reference values.
- The same four-fragment decomposition works across RHF, CCSD, VQE, ADAPT-VQE, and SQD solvers for chains from C4 to C26.
Where Pith is reading between the lines
- The fixed four-calculation count could extend to other linear chain molecules if similar radical symmetry holds.
- Hardware runs on fragments suggest the method lowers the barrier for applying current quantum devices to molecules larger than those feasible today.
- If the radical-fragment choice avoids the need for electrostatic embedding, the same pattern may simplify other fragmentation schemes that currently rely on capping and embedding.
Load-bearing premise
That two-body interactions of isolated radical fragments are enough to reconstruct accurate total energies without higher-order corrections or embedding terms for linear alkanes.
What would settle it
A direct full-molecule calculation for an alkane such as C30H62 that deviates from the MBE2 energy by more than chemical accuracy using any of the tested solvers.
Figures


read the original abstract
We introduce a radical-fragment many-body expansion at the two-body level (MBE2) for quantum chemistry of linear alkanes. Instead of heterolytic bond cleavage with hydrogen capping atoms and electrostatic embedding like in Fragment Molecular Orbital (FMO), we perform homolytic C-C bond cleavage to produce open-shell radical fragments (CH3, CH2) treated with restricted open-shell Hartree-Fock (ROHF) in isolation. The two-body MBE2 assembly formula reconstructs total alkane energies from only four unique fragment calculations regardless of chain length, reducing the maximum qubit requirement. We benchmark this framework against five energy solvers (RHF, CCSD, VQE, ADAPT-VQE, and SQD) across 11 linear alkanes from butane (C4H10) to hexacosane (C26H54). The MBE2 decomposition achieves a 12.3x qubit reduction for C26H54 (from 368 to 30 qubits) and a 12.8x reduction in unique calculations via symmetry exploitation. MBE2-VQE and MBE2-SQD (executed on IBM quantum hardware) closely track their respective classical MBE2 references, demonstrating that fragmentation-based quantum chemistry is viable for scaling quantum solvers to large molecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a radical-fragment many-body expansion at the two-body level (MBE2) for linear alkanes, using homolytic C-C bond cleavage to generate open-shell radical fragments (CH3, CH2) treated in isolation with ROHF. An assembly formula is claimed to reconstruct total energies from only four unique fragment calculations independent of chain length. The approach is benchmarked on 11 alkanes (C4H10 to C26H54) using RHF, CCSD, VQE, ADAPT-VQE, and SQD solvers, reporting a 12.3x qubit reduction for C26H54 (368 to 30 qubits) and close tracking of MBE2-VQE/MBE2-SQD (on IBM hardware) to classical MBE2 references.
Significance. If the two-body radical-fragment MBE2 is shown to accurately reproduce full-molecule energies, the method would offer a concrete route to reduce qubit counts and unique circuit evaluations for quantum chemistry on linear systems, potentially extending the reach of near-term quantum solvers without embedding or higher-order corrections.
major comments (2)
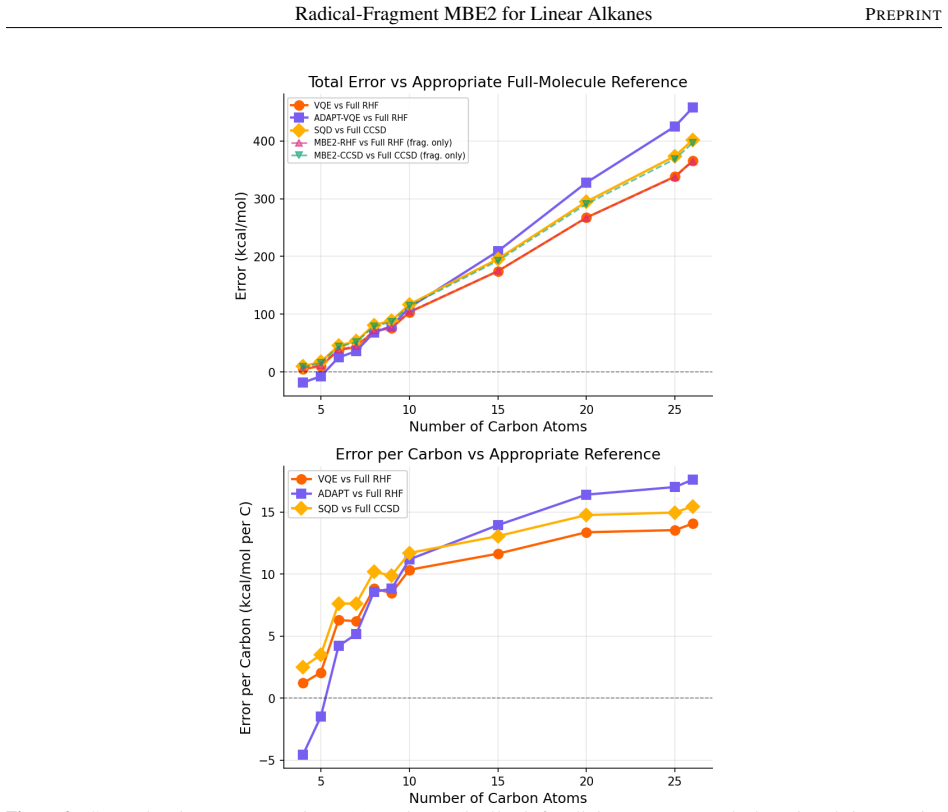
- [Abstract] Abstract: the central reconstruction claim requires that the two-body MBE2 formula (homolytic cleavage, isolated ROHF fragments, no embedding) accurately recovers total alkane energies, yet the reported benchmarks compare only MBE2-VQE and MBE2-SQD to classical MBE2 references; no errors versus non-fragmented full-molecule RHF or CCSD are supplied for any of the 11 alkanes, leaving the sufficiency of the two-body truncation unverified.
- [Abstract] Abstract: the qubit-reduction and symmetry-exploitation claims (12.3x and 12.8x for C26H54) rest on the assembly formula using only four unique fragments, but without an explicit equation for the MBE2 energy expression or a demonstration that higher-order terms remain negligible for linear alkanes, the scaling advantage cannot be assessed against full-molecule references.
Simulated Author's Rebuttal
We thank the referee for their detailed and constructive review. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central reconstruction claim requires that the two-body MBE2 formula (homolytic cleavage, isolated ROHF fragments, no embedding) accurately recovers total alkane energies, yet the reported benchmarks compare only MBE2-VQE and MBE2-SQD to classical MBE2 references; no errors versus non-fragmented full-molecule RHF or CCSD are supplied for any of the 11 alkanes, leaving the sufficiency of the two-body truncation unverified.
Authors: The manuscript's primary objective is to validate that quantum solvers applied to the radical fragments reproduce the classical MBE2 reference energies, thereby establishing the viability of the fragmentation strategy for quantum hardware. The two-body reconstruction formula itself is presented in the main text (Eq. 1) and is constructed to recover the total energy exactly within the MBE2 approximation for the chosen homolytic cleavage. Direct MBE2 vs. full-molecule errors are not reported because the focus is on the quantum-classical agreement on the fragments rather than re-benchmarking the classical MBE2 truncation. We agree this comparison would strengthen the abstract claim and will add a supplementary table of MBE2-RHF versus full RHF energies for the smaller alkanes (C4–C10) where full calculations remain tractable. revision: yes
-
Referee: [Abstract] Abstract: the qubit-reduction and symmetry-exploitation claims (12.3x and 12.8x for C26H54) rest on the assembly formula using only four unique fragments, but without an explicit equation for the MBE2 energy expression or a demonstration that higher-order terms remain negligible for linear alkanes, the scaling advantage cannot be assessed against full-molecule references.
Authors: Equation 1 in Section II explicitly states the MBE2 assembly: the total energy is recovered from only four unique open-shell fragment calculations (two distinct CH3 radicals and two distinct CH2 radicals) with appropriate coefficients independent of chain length. The qubit counts (368 → 30 for C26H54) follow directly from solving these fixed-size fragments rather than the full molecule. For linear alkanes the two-body truncation is justified by the localized C–C interactions; the consistent agreement between MBE2-VQE/SQD and classical MBE2 across eleven chain lengths provides indirect support that omitted higher-order terms do not grow with length. We will add a brief clarifying sentence in the revised abstract and a footnote referencing Eq. 1 to make the scaling basis unambiguous. revision: partial
Circularity Check
No circularity: derivation is self-contained by explicit construction of MBE2
full rationale
The paper explicitly defines the radical-fragment MBE2 procedure (homolytic cleavage to CH3/CH2 radicals, isolated ROHF, two-body assembly from four unique fragments) and applies it to linear alkanes. It then compares quantum solvers (VQE, SQD) directly to classical MBE2 references computed on the identical fragments. Qubit and calculation reductions follow immediately from the fragmentation definition itself. No parameters are fitted to a data subset and renamed as predictions, no self-citations justify load-bearing uniqueness or ansatze, and no equations reduce to their inputs by construction. The reported tracking of references is therefore a direct consequence of solving the same defined subproblems, rendering the chain self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard quantum chemistry solvers (RHF, CCSD, VQE) applied to isolated radical fragments yield energies that combine correctly via the MBE2 formula.
Reference graph
Works this paper leans on
-
[1]
Fragment molecular orbital method: an approximate computational method for large molecules,
K. Kitaura et al., “Fragment molecular orbital method: an approximate computational method for large molecules,” Chem. Phys. Lett., vol. 313, pp. 701–706, 1999. 11 Radical-Fragment MBE2 for Linear AlkanesPREPRINT
1999
-
[2]
Extending the Power of Quantum Chemistry to Large Systems with the Fragment Molecular Orbital Method,
D. G. Fedorov and K. Kitaura, “Extending the Power of Quantum Chemistry to Large Systems with the Fragment Molecular Orbital Method,”J. Phys. Chem. A, vol. 111, pp. 6904–6914, 2007
2007
-
[3]
The fragment molecular orbital method: theoretical development, implementation in GAMESS, and applications,
D. G. Fedorov, “The fragment molecular orbital method: theoretical development, implementation in GAMESS, and applications,”WIREs Comput. Mol. Sci., vol. 7, e1322, 2017
2017
-
[4]
A variational eigenvalue solver on a photonic quantum processor,
A. Peruzzo et al., “A variational eigenvalue solver on a photonic quantum processor,”Nat. Commun., vol. 5, 4213, 2014
2014
-
[5]
An adaptive variational algorithm for exact molecular simulations on a quantum computer,
H. R. Grimsley et al., “An adaptive variational algorithm for exact molecular simulations on a quantum computer,” Nat. Commun., vol. 10, 3007, 2019
2019
-
[6]
Chemistry Beyond Exact Solutions on a Quantum-Centric Supercomputer,
J. Robledo-Moreno, M. Motta, H. Haas et al., “Chemistry Beyond Exact Solutions on a Quantum-Centric Supercomputer,”Sci. Adv., 2025
2025
-
[7]
Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry in the age of quantum computing,
H. Lim et al., “Fragment molecular orbital-based variational quantum eigensolver for quantum chemistry in the age of quantum computing,”Sci. Rep., vol. 14, 2422, 2024
2024
-
[8]
Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,
A. Kandala et al., “Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,” Nature, vol. 549, pp. 242–246, 2017
2017
-
[9]
The theory of variational hybrid quantum-classical algorithms,
J. R. McClean et al., “The theory of variational hybrid quantum-classical algorithms,”New J. Phys., vol. 18, 023023, 2016
2016
-
[10]
Szabo and N
A. Szabo and N. S. Ostlund,Modern Quantum Chemistry. Dover Publications, 1996
1996
-
[11]
PySCF: the Python-based simulations of chemistry framework,
Q. Sun et al., “PySCF: the Python-based simulations of chemistry framework,”WIREs Comput. Mol. Sci., vol. 8, e1340, 2018
2018
-
[12]
Über das Paulische Äquivalenzverbot,
P. Jordan and E. Wigner, “Über das Paulische Äquivalenzverbot,”Z. Phys., vol. 47, pp. 631–651, 1928
1928
-
[13]
Cholesky Decomposition Techniques in Electronic Structure Theory,
F. Aquilante et al., “Cholesky Decomposition Techniques in Electronic Structure Theory,” inLinear-Scaling Techniques in Computational Chemistry and Physics, Springer, 2011
2011
-
[14]
Implementation of the simultaneous perturbation algorithm for stochastic optimization,
J. C. Spall, “Implementation of the simultaneous perturbation algorithm for stochastic optimization,”IEEE Trans. Aerosp. Electron. Syst., vol. 34, pp. 817–823, 1998
1998
-
[15]
Generalized Unitary Coupled Cluster Wave functions for Quantum Computation,
J. Lee et al., “Generalized Unitary Coupled Cluster Wave functions for Quantum Computation,”J. Chem. Theory Comput., vol. 15, pp. 311–324, 2019
2019
-
[16]
Adaptive, problem-tailored variational quantum eigensolver mitigates rough parameter landscapes and barren plateaus,
H. R. Grimsley et al., “Adaptive, problem-tailored variational quantum eigensolver mitigates rough parameter landscapes and barren plateaus,”npj Quantum Inf., vol. 9, 19, 2023
2023
-
[17]
qubit-ADAPT-VQE: An adaptive algorithm for constructing hardware-efficient ansätze on a quantum processor,
H. L. Tang et al., “qubit-ADAPT-VQE: An adaptive algorithm for constructing hardware-efficient ansätze on a quantum processor,”PRX Quantum, vol. 2, 020310, 2021
2021
-
[18]
Extension of the fragment molecular orbital method to treat large open-shell systems in solution,
D. G. Fedorov et al., “Extension of the fragment molecular orbital method to treat large open-shell systems in solution,”Chem. Phys. Lett., vol. 649, pp. 60–65, 2016
2016
-
[19]
Qiskit: An Open-Source Framework for Quantum Computing,
Qiskit Development Team, “Qiskit: An Open-Source Framework for Quantum Computing,” 2024
2024
-
[20]
Quantum HF/DFT-embedding algorithms for electronic structure calculations,
M. Rossmannek et al., “Quantum HF/DFT-embedding algorithms for electronic structure calculations,”J. Chem. Phys., vol. 154, 114105, 2021. 12
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.