Many-body benchmarking of DFT local-registry energetics in bilayer InSe
Pith reviewed 2026-07-03 10:19 UTC · model grok-4.3
The pith
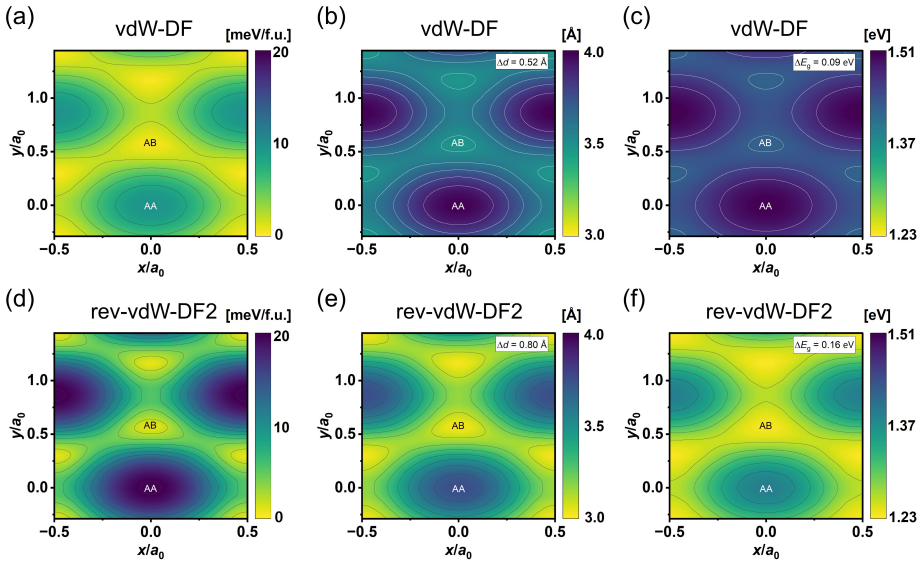
DMC shows InSe bilayer stackings differ by up to 60 meV per formula unit while DFT finds them nearly degenerate.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
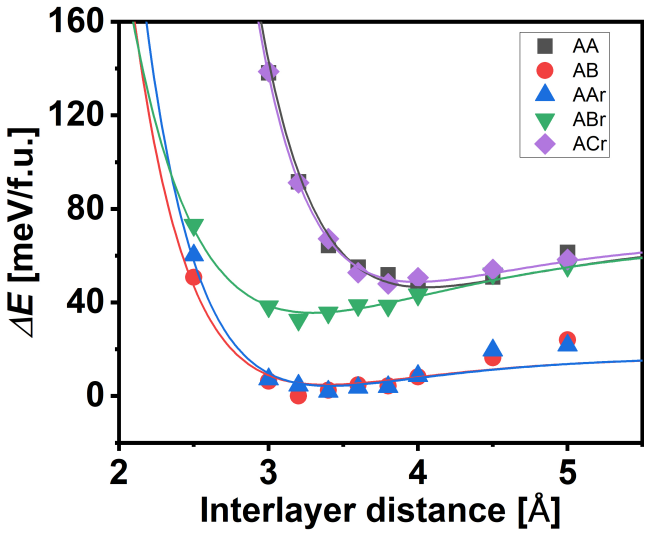
DMC separates AB, AAr, and ABr stackings by 8(5) and 41(4) meV/f.u., while the energy difference between the most stable and least stable registries reaches 60(7) meV/f.u.. These large energy separations show that the stacking energetics are not determined by the interfacial atomic motif alone but depend on the full registry and its associated many-body electronic response. More broadly, these results show that DFT-based moiré models can substantially underestimate local stacking-energy corrugation, with direct consequences for predicted structural relaxation, domain formation, and electronic reconstruction in twisted layered materials.
What carries the argument
Diffusion quantum Monte Carlo (DMC) applied to the three high-symmetry stackings of bilayer InSe to extract registry-dependent total energies beyond DFT.
If this is right
- Twisted InSe bilayers will exhibit stronger structural relaxation and larger domain sizes than DFT moiré models predict.
- Electronic reconstruction at domain walls will be more pronounced because of the deeper local energy wells.
- Similar underestimation of stacking corrugation is expected in other van der Waals bilayers when only DFT is used.
Where Pith is reading between the lines
- Models that fit moiré potentials from DFT alone may need systematic corrections derived from many-body methods for quantitative predictions of twist-angle-dependent properties.
- Local stacking energies could be measured by combining atomic-resolution imaging with spectroscopy to test the DMC scale directly.
- The many-body contribution may also affect phonon modes or excitonic binding that depend on interlayer registry.
Load-bearing premise
The three chosen high-symmetry stackings represent the dominant local registries that appear in a continuously twisted bilayer and that the DMC energies for these discrete cells capture the main many-body contributions without large finite-size or convergence errors.
What would settle it
A converged DMC or higher-level calculation on larger supercells that brings the registry energy spread below roughly 10 meV per formula unit, or an experimental probe that measures stacking-energy differences near the DFT values, would falsify the central claim.
Figures

read the original abstract
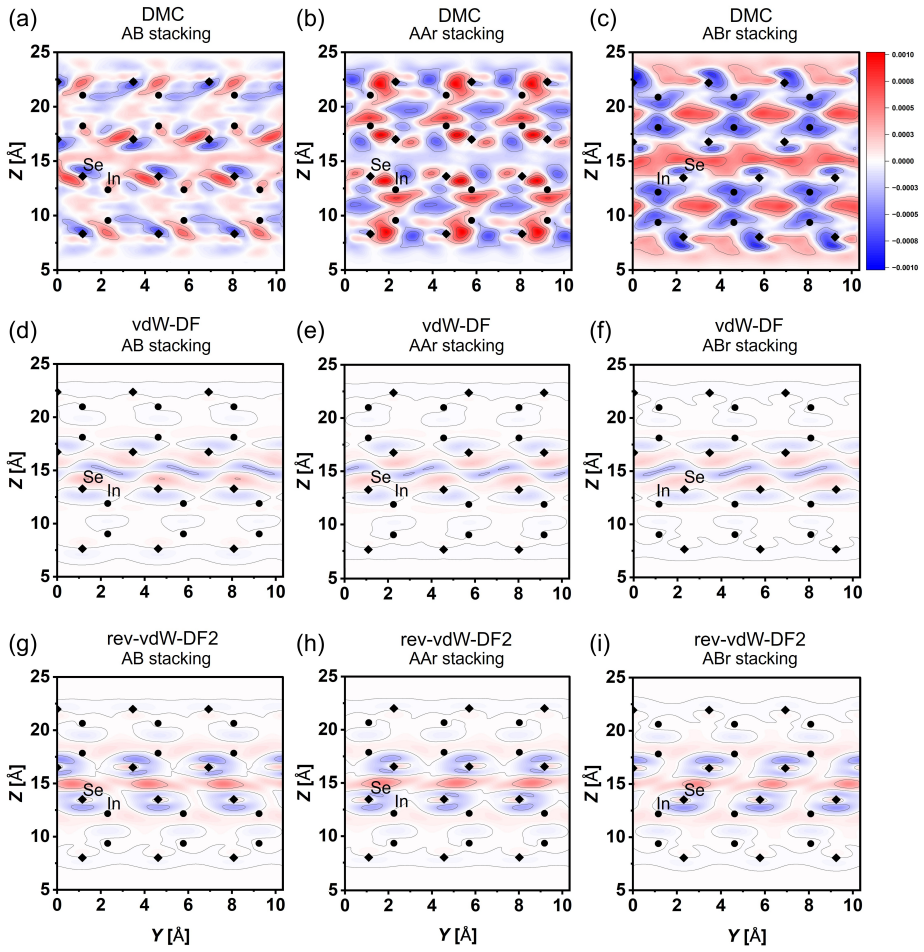
Density functional theory (DFT) is widely used to model twisted bilayers, but the accuracy of the local stacking energetics underlying such models remains uncertain. Here, we benchmark the local-registry landscape of bilayer InSe using diffusion quantum Monte Carlo (DMC). DFT predicts that AB, AAr, and ABr stackings, which share the same interfacial Se registry, are nearly degenerate within 1.5 meV/f.u. and exhibit nearly indistinguishable DFT charge-density responses. DMC instead separates these stackings by 8(5) and 41(4) meV/f.u., while the energy difference between the most stable and least stable registries reaches 60(7) meV/f.u.. These large energy separations show that the stacking energetics are not determined by the interfacial atomic motif alone but depend on the full registry and its associated many-body electronic response. More broadly, these results show that DFT-based moir\'e models can substantially underestimate local stacking-energy corrugation, with direct consequences for predicted structural relaxation, domain formation, and electronic reconstruction in twisted layered materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that DFT finds AB, AAr, and ABr stackings in bilayer InSe nearly degenerate (within 1.5 meV/f.u.) with similar charge densities, while DMC calculations separate them by 8(5) and 41(4) meV/f.u. with a maximum range of 60(7) meV/f.u.; this demonstrates that local stacking energetics depend on the full registry and many-body electronic response rather than the shared interfacial Se motif alone, implying that DFT-based moiré models substantially underestimate corrugation and its consequences for relaxation and reconstruction.
Significance. If the DMC energy differences hold after addressing statistical and convergence issues, the result provides a valuable many-body benchmark for twisted bilayer energetics in InSe, highlighting limitations of DFT local-registry approximations with direct implications for moiré modeling in layered materials. The direct DMC vs. DFT comparison on identical configurations is a strength.
major comments (2)
- [Abstract] Abstract and results: The reported DMC energy difference of 8(5) meV/f.u. between AB and AAr stackings is only ~1.6σ from zero. This marginal separation undermines the central claim that DMC 'separates these stackings' and that energetics 'are not determined by the interfacial atomic motif alone,' since the headline assertion requires all three registries to be distinctly resolved beyond the DFT near-degeneracy.
- [Methods] Methods/results: No convergence tests, system-size scaling, or detailed methodology (e.g., twist-angle handling, finite-size corrections, or DMC parameters) are provided for the InSe bilayer. This directly threatens verification of the many-body response conclusion and the weakest assumption that the discrete high-symmetry configurations capture the dominant effects without specific convergence errors.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for highlighting these important points regarding statistical significance and methodological details. We respond to each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract and results: The reported DMC energy difference of 8(5) meV/f.u. between AB and AAr stackings is only ~1.6σ from zero. This marginal separation undermines the central claim that DMC 'separates these stackings' and that energetics 'are not determined by the interfacial atomic motif alone,' since the headline assertion requires all three registries to be distinctly resolved beyond the DFT near-degeneracy.
Authors: We agree that the 8(5) meV/f.u. difference is only ~1.6σ and is therefore marginal, which weakens the assertion of clear separation specifically between AB and AAr. The larger 41(4) meV/f.u. separation and 60(7) meV/f.u. overall range still demonstrate that the three stackings are not all near-degenerate within the DFT value of 1.5 meV/f.u., supporting the broader conclusion that energetics depend on the full registry. We will revise the abstract and main text to qualify the AB-AAr result explicitly in terms of its statistical significance while retaining the claim for the resolved separations. revision: partial
-
Referee: [Methods] Methods/results: No convergence tests, system-size scaling, or detailed methodology (e.g., twist-angle handling, finite-size corrections, or DMC parameters) are provided for the InSe bilayer. This directly threatens verification of the many-body response conclusion and the weakest assumption that the discrete high-symmetry configurations capture the dominant effects without specific convergence errors.
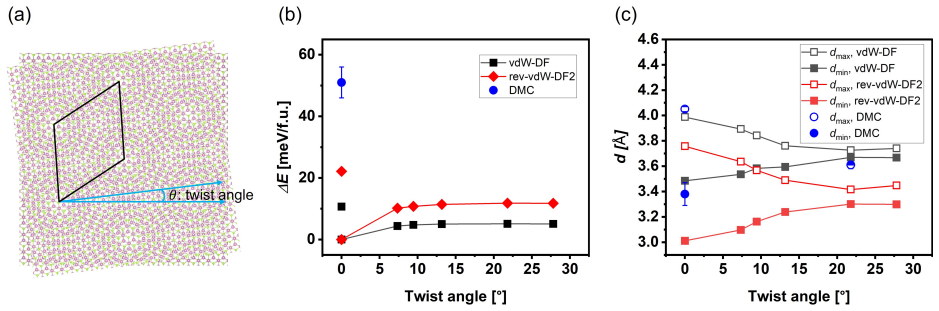
Authors: The submitted manuscript indeed omits explicit convergence tests, system-size scaling, finite-size corrections, and full DMC parameter details. We will add these in a revised methods section, including justification that the high-symmetry commensurate configurations are appropriate for isolating local-registry effects. Since the study addresses fixed bilayer stackings rather than incommensurate twisted structures, twist-angle handling does not apply and will be clarified as such. revision: yes
Circularity Check
No circularity: energies are direct outputs of independent DMC/DFT runs on fixed configurations.
full rationale
The paper computes total energies for three high-symmetry stackings via DMC and DFT on explicitly defined atomic geometries. The reported differences (8(5), 41(4), 60(7) meV/f.u.) are numerical results of those runs, not quantities obtained by fitting a parameter to a subset of the same data, by algebraic reduction to an input, or by a self-citation chain that itself depends on the target result. No equations or ansätze are introduced that would make any claimed separation equivalent to its own inputs by construction. The central claim therefore rests on external, falsifiable simulation outputs rather than on any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Born-Oppenheimer approximation and standard pseudopotential treatment of core electrons are valid for the InSe bilayer.
Reference graph
Works this paper leans on
-
[1]
Yoo, H. et al. Atomic and electronic reconstruction at the van der Waals interface in twisted bilayer graphene. Nat. Mater.18, 448 (2019)
work page 2019
-
[2]
Carr, S., Fang, S. & Kaxiras, E. Electronic-structure methods for twisted moir´ e layers. Nat. Rev. Mater.5, 748 (2020). 19
work page 2020
-
[3]
Zhou, J. et al. InSe monolayer: synthesis, structure and ultra-high second-harmonic genera- tion. 2D Mater.5, 025019 (2018)
work page 2018
-
[4]
Hilse, M. et al. Mixed polytype/polymorph formation in InSe films grown by molecular beam epitaxy on GaAs(111)B. npj 2D Mater. Appl.9, 19 (2025)
work page 2025
-
[5]
Song, S. et al. Indium selenides for next-generation low-power computing devices. Nat. Rev. Electr. Eng.3, 185 (2026)
work page 2026
-
[6]
Mudd, G. W. et al. Tuning the bandgap of exfoliated InSe nanosheets by quantum confinement. Adv. Mater.25, 5714 (2013)
work page 2013
-
[7]
Sun, Y. et al. InSe: a two-dimensional material with strong interlayer coupling. Nanoscale 10, 7991 (2018)
work page 2018
-
[8]
Yao, X. & Zhang, X. Electronic structures of twisted bilayer InSe/InSe and heterobilayer graphene/InSe. ACS Omega6, 13426 (2021)
work page 2021
-
[9]
Pike, N. A. et al. Understanding the origin and implication of the indirect-to-direct bandgap transition in multilayer InSe. J. Phys. Chem. C128, 7957 (2024)
work page 2024
-
[10]
Magorrian, S. J., Siddiqui, A. & Hine, N. D. Strong atomic reconstruction in twisted bilayers of highly flexible InSe: Machine-learned interatomic potential and continuum model approaches. Phys. Rev. Mater.9, 014004 (2025)
work page 2025
-
[11]
Foulkes, W. M. C., Mitas, L., Needs, R. J. & Rajagopal, G. Quantum monte carlo simulations of solids. Rev. Mod. Phys.73, 33 (2001)
work page 2001
-
[12]
Han, L. et al. Strong interlayer interaction in two-dimensional layered PtTe 2. J. Solid State Chem.305, 122657 (2022)
work page 2022
-
[13]
Ahn, J., Kang, S.-H., Yoon, M. & Krogel, J. T. Exploring interlayer coupling in the twisted bilayer PtTe2. Phys. Rev. Res.6, 033177 (2024)
work page 2024
-
[14]
Li, J. et al. Layer-dependent band gaps of platinum dichalcogenides. ACS Nano15, 13249 (2021)
work page 2021
-
[15]
Ahn, J. et al. Stacking polymorphism of PtSe 2: its implication to layer-dependent metal- insulator transitions. npj 2D Mater. Appl.9, 34 (2025)
work page 2025
-
[16]
Politano, A. et al. Indium selenide: an insight into electronic band structure and surface excitations. Sci. Rep.7, 3445 (2017)
work page 2017
-
[17]
Shin, H. et al. Optimized structure and electronic band gap of monolayer GeSe from quantum Monte Carlo methods. Phys. Rev. Mater.5, 024002 (2021). 20
work page 2021
-
[18]
Kim, J. et al. QMCPACK : An open source ab initio Quantum Monte Carlo package for the electronic structure of atoms, molecules, and solids. J. Phys.: Condens. Matter30, 195901 (2018)
work page 2018
-
[19]
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.77, 3865 (1996)
work page 1996
-
[20]
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter21, 395502 (2009)
work page 2009
- [21]
-
[22]
Casula, M., Moroni, S., Sorella, S. & Filippi, C. Size-consistent variational approaches to nonlocal pseudopotentials: Standard and lattice regularized diffusion Monte Carlo methods revisited. J. Chem. Phys.132, 154113 (2010)
work page 2010
-
[23]
Lin, C., Zong, F. H. & Ceperley, D. M. Twist-averaged boundary conditions in continuum quantum Monte Carlo algorithms. Phys. Rev. E64, 016702 (2001)
work page 2001
-
[24]
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B47, 558 (1993)
work page 1993
-
[25]
Kresse, G. & Furthm¨ uller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B54, 11169 (1996)
work page 1996
-
[26]
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B59, 1758 (1999)
work page 1999
-
[27]
Dion, M., Rydberg, H., Schr¨ oder, E., Langreth, D. C. & Lundqvist, B. I. Van der Waals density functional for general geometries. Phys. Rev. Lett.92, 246401 (2004)
work page 2004
-
[28]
Lee, K., Murray, ´E. D., Kong, L., Lundqvist, B. I. & Langreth, D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B82, 081101 (2010)
work page 2010
-
[29]
van der Waals density functional made accurate
Hamada, I. van der Waals density functional made accurate. Phys. Rev. B89, 121103 (2014)
work page 2014
-
[30]
Klimeˇ s, J., Bowler, D. R. & Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B83, 195131 (2011)
work page 2011
-
[31]
Klimeˇ s, J., Bowler, D. R. & Michaelides, A. Chemical accuracy for the van der Waals density functional. J. Phys.: Condens. Matter22, 022201 (2010)
work page 2010
-
[32]
Peng, H., Yang, Z.-H., Perdew, J. P. & Sun, J. Versatile van der Waals density functional based on a meta-generalized gradient approximation. Phys. Rev. X6, 041005 (2016). 21
work page 2016
-
[33]
Ning, J. et al. Workhorse minimally empirical dispersion-corrected density functional with tests for weakly bound systems: r 2SCAN+rVV10. Phys. Rev. B106, 075422 (2022)
work page 2022
-
[34]
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys.132, 154104 (2010)
work page 2010
-
[35]
Tkatchenko, A., DiStasio Jr, R. A., Car, R. & Scheffler, M. Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett.108, 236402 (2012). 22
work page 2012
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.