Theory of Cation Solvation in the Helmholtz Layer of Li-ion Battery Electrolytes
Pith reviewed 2026-05-23 01:18 UTC · model grok-4.3
The pith
A theory shows Li+ solvent binding probabilities in the Helmholtz layer equal those in the bulk.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
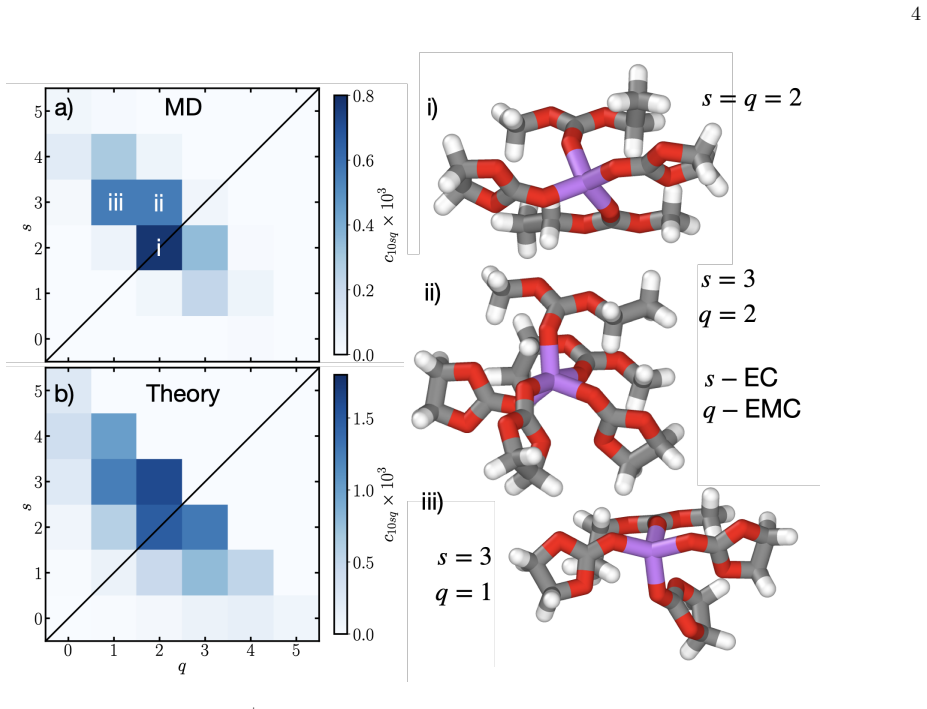
By solving a simplified version of the developed theory of cation solvation in the Helmholtz layer, the probability of Li+ binding to each solvent remains equal to the bulk probability, suggesting bulk solvation environments are a reasonable starting point for understanding new battery electrolytes.
What carries the argument
Statistical theory of Li+ binding to solvents where the Helmholtz layer effect is captured by an apparent reduction in the number of binding sites, parameterized from bulk MD simulations.
If this is right
- The theory accurately reproduces solvation environments from MD in bulk and diffuse EDL as a function of surface charge.
- The dominant interface effect in the Helmholtz layer is an apparent reduction in binding sites between Li+ and solvents.
- Bulk solvation probabilities provide a good proxy for predicting reactions at the electrode.
- The formalism allows prediction of Helmholtz layer solvation from bulk data alone to inform SEI formation.
Where Pith is reading between the lines
- If the binding probabilities are unchanged, screening new electrolyte compositions can rely primarily on bulk simulations rather than expensive interface models.
- This approach might extend to other battery chemistries where solvation dictates interfacial reactions.
- Testing the theory with different electrode materials could reveal if the binding site reduction is universal or material-specific.
Load-bearing premise
The dominant change near the electrode can be modeled solely as a reduction in the number of Li+ solvent binding sites, with parameterization from bulk data absorbing all other interface effects.
What would settle it
Running atomistic MD simulations of the full Helmholtz layer and checking whether the observed Li+ solvent binding probabilities deviate from the bulk values predicted by the simplified theory.
Figures



read the original abstract
The solvation environments of Li$^+$ in conventional non-aqueous battery electrolytes, such as LiPF$_6$ in mixtures of ethylene carbaronate (EC) and ethyl methyl carbonate (EMC), are often used to rationalize the transport properties of electrolytes and solid electrolyte interphase (SEI) formation. In the SEI, the solvation environments in the compact electrical double layer (EDL) next to the electrode, also known as the Helmholtz layer, determine (partially) what species can react to form the SEI, with bulk solvation environments often being used as a proxy. Here we develop and test a theory of cation solvation in the Helmholtz layer of non-aqueous Li-ion battery electrolytes. First, we validate the theory against bulk and diffuse EDL atomistic molecular dynamics (MD) simulations of LiPF$_6$ EC/EMC mixtures as a function of surface charge, where we find the theory can capture the solvation environments well. Next we turn to the Helmholtz layer, where we find that the main effect of the solvation structures next to the electrode is an apparent reduction in the number of binding sites between Li$^+$ and the solvents, again where we find good agreement with our developed theory. Finally, by solving a simplified version of the theory, we find that the probability of Li$^+$ binding to each solvent remains equal to the bulk probability, suggesting that the bulk solvation environments are a reasonable place to start when understanding new battery electrolytes. Our developed formalism can be parameterized from bulk MD simulations and used to predict the solvation environments in the Helmholtz layer, which can be used to determine what could react and form the SEI.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops a statistical-mechanical theory for Li+ solvation in the Helmholtz layer of non-aqueous Li-ion battery electrolytes (LiPF6 in EC/EMC mixtures). It validates the theory against MD for bulk and diffuse EDL regimes, identifies an apparent reduction in Li+-solvent binding sites as the dominant Helmholtz-layer effect, and solves a simplified version of the theory to conclude that Li+ binding probabilities to each solvent remain equal to their bulk values, implying that bulk solvation environments are a reasonable proxy for SEI-relevant species.
Significance. If the central result holds, the formalism offers a route to predict compact-layer solvation from bulk MD data alone, which would simplify screening of electrolyte mixtures for SEI formation. The explicit finding that binding probabilities are insensitive to the interface under the reduced-site model provides a concrete, falsifiable claim that could guide both simulation and experiment in battery electrolyte design.
major comments (3)
- [simplified-solution section] The claim that binding probabilities remain bulk-like rests on solving a simplified version of the theory (abstract and the section presenting the simplified solution). No explicit check is provided that the neglected position-dependent electrostatic and density terms remain small once the surface charge places Li+ in the compact layer; if these terms shift chemical potentials differently for EC versus EMC, the probabilities would deviate even while the reduced-site model fits average coordination numbers.
- [validation sections] Validation against MD is described only qualitatively as 'good agreement' for bulk, diffuse EDL, and Helmholtz layer (abstract). No quantitative metrics (RMSE, R², error bars on coordination numbers, or details on how the single binding-site reduction factor was obtained from bulk MD) are reported, leaving the load-bearing claim of predictive power for the Helmholtz layer uninspectable.
- [theory-development and Helmholtz-layer sections] The central modeling step assumes that all Helmholtz-layer effects are captured by a single apparent reduction in binding sites, with all other interface contributions absorbed into this parameter when the theory is fit to bulk MD. This assumption is load-bearing for the conclusion that bulk probabilities are unchanged; an explicit test (e.g., comparing full vs. reduced-site solutions at finite surface charge) is needed to confirm the omitted terms do not alter relative binding free energies.
minor comments (2)
- [abstract] Abstract contains the typo 'carbaronate' (should be 'carbonate').
- [theory section] Notation for the binding-site reduction factor and its relation to the full partition function should be clarified to avoid ambiguity when the simplified solution is presented.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments. We have revised the manuscript to incorporate quantitative validation metrics and an explicit test of the reduced-site model assumptions. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [simplified-solution section] The claim that binding probabilities remain bulk-like rests on solving a simplified version of the theory (abstract and the section presenting the simplified solution). No explicit check is provided that the neglected position-dependent electrostatic and density terms remain small once the surface charge places Li+ in the compact layer; if these terms shift chemical potentials differently for EC versus EMC, the probabilities would deviate even while the reduced-site model fits average coordination numbers.
Authors: We agree that an explicit verification strengthens the argument. In the revised manuscript we have added a direct comparison of the full theory versus the reduced-site model at surface charges placing Li+ in the Helmholtz layer. The relative binding probabilities for EC and EMC remain equal to bulk values within numerical precision, indicating that the neglected position-dependent terms do not differentially shift the chemical potentials under the conditions examined. revision: yes
-
Referee: [validation sections] Validation against MD is described only qualitatively as 'good agreement' for bulk, diffuse EDL, and Helmholtz layer (abstract). No quantitative metrics (RMSE, R², error bars on coordination numbers, or details on how the single binding-site reduction factor was obtained from bulk MD) are reported, leaving the load-bearing claim of predictive power for the Helmholtz layer uninspectable.
Authors: We accept that quantitative metrics improve inspectability. The revised manuscript now reports RMSE and R² values together with error bars on all coordination-number comparisons for bulk, diffuse EDL, and Helmholtz-layer regimes. The methods section additionally details the fitting procedure used to extract the single binding-site reduction factor from bulk MD coordination numbers. revision: yes
-
Referee: [theory-development and Helmholtz-layer sections] The central modeling step assumes that all Helmholtz-layer effects are captured by a single apparent reduction in binding sites, with all other interface contributions absorbed into this parameter when the theory is fit to bulk MD. This assumption is load-bearing for the conclusion that bulk probabilities are unchanged; an explicit test (e.g., comparing full vs. reduced-site solutions at finite surface charge) is needed to confirm the omitted terms do not alter relative binding free energies.
Authors: The assumption is central, and we have performed the requested explicit test. The revised text now compares full-theory and reduced-site solutions at finite surface charge; the comparison shows that relative binding free energies are unaffected once the site-reduction parameter is fixed from bulk data, thereby supporting the conclusion that binding probabilities remain bulk-like. revision: yes
Circularity Check
Simplified Helmholtz solution recovers bulk binding probabilities by construction from bulk-MD parameterization
specific steps
-
fitted input called prediction
[Abstract (final paragraph)]
"Finally, by solving a simplified version of the theory, we find that the probability of Li+ binding to each solvent remains equal to the bulk probability... Our developed formalism can be parameterized from bulk MD simulations and used to predict the solvation environments in the Helmholtz layer"
The theory is parameterized from bulk MD by a single effective reduction in binding sites that absorbs all interface effects. The simplified solution that recovers exactly the bulk binding probabilities is therefore equivalent to the fitted bulk statistics by construction; no independent calculation of the neglected electrostatic or density terms is performed.
full rationale
The paper explicitly states that its formalism is parameterized from bulk MD by introducing a single effective reduction in Li+-solvent binding sites that captures all Helmholtz-layer effects. Solving the resulting simplified theory then yields unchanged binding probabilities equal to the bulk values. This outcome follows directly from the parameterization choice and the omission of position-dependent terms rather than from an independent derivation of those terms. The central claim that bulk solvation environments remain a reasonable proxy therefore reduces to the fitted input.
Axiom & Free-Parameter Ledger
free parameters (1)
- apparent binding-site reduction factor
axioms (1)
- domain assumption Cation solvation can be represented as statistical occupancy of a fixed number of binding sites by solvent molecules
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the main effect of the solvation structures next to the electrode is an apparent reduction in the number of binding sites between Li+ and the solvents... by solving a simplified version of the theory, we find that the probability of Li+ binding to each solvent remains equal to the bulk probability
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Wlmsq = (f+l − l)!(f−m − m)! / l!m!s!q!(f+l − l − m − s − q + 1)! ... λi = e−βΔf+i ... mass action laws
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson, and G. Ceder, Chem. Rev. 121, 1623 (2021)
work page 2021
-
[2]
M. Li, C. Wang, Z. Chen, K. Xu, and J. Lu, Chem. Rev. 120, 6783 (2020)
work page 2020
-
[3]
W. Li, E. M. Erickson, and A. Manthiram, Nat. Energy 5, 26 (2020)
work page 2020
-
[4]
H. Wang, Z. Yu, X. Kong, S. C. Kim, D. T. Boyle, J. Qin, Z. Bao, and Y. Cui, Joule 6, 588 (2022)
work page 2022
-
[5]
Y. S. Meng, V. Srinivasan, and K. Xu, Science 378, 1065 (2022)
work page 2022
- [6]
- [7]
-
[8]
Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, et al. , Nat. Energy 5, 526 (2020)
work page 2020
-
[9]
Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, et al. , Nat. Energy 7, 94 (2022)
work page 2022
-
[10]
K. Xu, Y. Lam, S. S. Zhang, T. R. Jow, and T. B. Curtis, J. Phys. Chem. C 111, 7411 (2007)
work page 2007
-
[11]
A. von Wald Cresce, O. Borodin, and K. Xu, J. Phys. Chem. C 116, 26111 (2012)
work page 2012
-
[12]
Z. Li, O. Borodin, G. D. Smith, and D. Bedrov, J. Phys. Chem. B 119, 3085 (2015)
work page 2015
-
[13]
Z. Piao, R. Gao, Y. Liu, G. Zhou, and H.-M. Cheng, Adv.Mater. , 2206009 (2023)
work page 2023
-
[14]
Q. Wu, M. T. McDowell, and Y. Qi, JACS 145, 2473 (2023)
work page 2023
-
[15]
Z. A. Goodwin, M. McEldrew, B. Kozinsky, and M. Z. Bazant, PRX Energy 2, 013007 (2023)
work page 2023
- [16]
- [17]
- [18]
-
[19]
O. Borodin, J. Self, K. A. Persson, C. Wang, and K. Xu, Joule 4, 69 (2020)
work page 2020
- [20]
-
[21]
A. V. Cresce, S. M. Russell, O. Borodin, J. A. Allen, M. A. Schroeder, M. Dai, J. Peng, M. P. Gobet, S. G. Greenbaum, R. E. Rogers, and K. Xu, Phys. Chem. Chem. Phys. 19, 574 (2017)
work page 2017
-
[22]
D. M. Seo, O. Borodin, S.-D. Han, Q. Ly, P. D. Boyle, and W. A. Henderson, J. Electrochem. Soc. 159, A553 (2012)
work page 2012
-
[23]
D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle, and W. A. Henderson, J. Electrochem. Soc. 159, A1489 (2012)
work page 2012
-
[24]
L. Yang, A. Xiao, and B. L. Lucht, J. Mol. Liq. 154, 131 (2010). 11
work page 2010
- [25]
-
[26]
O. O. Postupna, Y. V. Kolesnik, O. N. Kalugin, and O. V. Prezhdo, J. Phys. Chem. B 115, 14563 (2011)
work page 2011
-
[27]
I. Skarmoutsos, V. Ponnuchamy, V. Vetere, and S. Mossa, J. Phys. Chem. C 119, 4502 (2015)
work page 2015
-
[28]
O. Borodin, X. Ren, J. Vatamanu, A. von Wald Cresce, J. Knap, and K. Xu, Acc. Chem. Res. 50, 2886 (2017)
work page 2017
- [29]
- [30]
- [31]
- [32]
-
[33]
N. Piao, X. Ji, H. Xu, X. Fan, L. Chen, S. Liu, M. N. Garaga, S. G. Greenbaum, L. Wang, C. Wang, and X. He, Adv. Energy Mater. 10, 1903568 (2020)
work page 2020
-
[34]
T. Hou, K. D. Fong, J. Wang, and K. A. Persson, Chem. Sci. 12, 14740 (2021)
work page 2021
-
[35]
Y. Wu, A. Wang, Q. Hu, H. Liang, H. Xu, L. Wang, and X. He, ACS Cent. Sci. 8, 1290 (2022)
work page 2022
-
[36]
C. M. Efaw, Q. Wu, N. Gao, Y. Zhang, H. Zhu, K. Ger- ing, M. F. Hurley, H. Xiong, E. Hu, X. Cao, W. Xu, J.-G. Zhang, E. J. Dufek, J. Xiao, X.-Q. Yang, J. Liu, Y. Qi, and B. Li, Nat. Mater 22, 1531 (2024)
work page 2024
-
[37]
M. J. Hossain, Q. Wu, E. J. M. Bernardez, C. D. Quilty, A. C. Marschilok, E. S. Takeuchi, D. C. Bock, K. J. Takeuchi, and Y. Qi, J. Phys. Chem. Lett. 14, 7718 (2023)
work page 2023
-
[38]
J. Chen, X. Fan, Q. Li, H. Yang, M. R. Khoshi, Y. Xu, S. Hwang, L. Chen, X. Ji, C. Yang, et al. , Nat. Energy 5, 386 (2020)
work page 2020
-
[39]
S. T. Oyakhire, S.-L. Liao, S. B. Shuchi, M. S. Kim, S. C. Kim, Z. Yu, R. A. Vil´ a, P. E. Rudnicki, Y. Cui, and S. F. Bent, Nano Lett. 23, 7524 (2023)
work page 2023
-
[40]
Q. Wu and Y. Qi, Energy Environ. Sci. , https://doi.org/10.1039/D5EE00206K (2025)
-
[41]
M. V. Fedorov and A. A. Kornyshev, Chem. Rev. 114, 2978 (2014)
work page 2014
-
[42]
M. Z. Bazant, M. S. Kilic, B. Storey, and A. Ajdari, Advances in Colloid and Interface Science152, 48 (2009)
work page 2009
-
[43]
Z. A. Goodwin, G. Feng, and A. A. Kornyshev, Elec- trochimica Acta 225, 190 (2017)
work page 2017
-
[44]
N. Yao, X. Chen, Z.-H. Fu, and Q. Zhang, Chem. Rev. 122, 10970 (2022)
work page 2022
-
[45]
F. Ren, Y. Wu, W. Zuo, W. Zhao, S. Pan, H. Lin, H. Yu, J. Lin, M. Lin, X. Yao, T. Brezesinski, Z. Gong, and Y. Yang, Energy Environ. Sci 17, 2743 (2024)
work page 2024
-
[46]
I.-B. Magd˘ au, D. J. Arismendi-Arrieta, H. E. Smith, C. P. Grey, K. Hermansson, and G. Cs´ anyi, 10.26434/chemrxiv-2022-l4tb9 (2022)
- [47]
-
[48]
Z. A. H. Goodwin, M. B. Wenny, J. H. Yang, A. Ce- pellotti, J. Ding, K. Bystrom, B. R. Duschatko, A. Jo- hansson, L. Sun, S. Batzner, A. Musaelian, J. A. Mason, B. Kozinsky, and N. Molinari, J. Phys. Chem. Lett. 15, 7539 (2024)
work page 2024
-
[49]
X. Xie, E. W. C. Spotte-Smith, M. Wen, H. D. Patel, S. M. Blau, and K. A. Persson, J. Am. Chem. Soc. 143, 13245 (2021)
work page 2021
-
[50]
E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau, and K. A. Persson, ACS Energy Lett. 7, 1446 (2022)
work page 2022
-
[51]
M. McEldrew, Z. A. H. Goodwin, S. Bi, M. Z. Bazant, and A. A. Kornyshev, J. Chem. Phys. 152, 234506 (2020)
work page 2020
-
[52]
M. McEldrew, Z. A. H. Goodwin, H. Zhao, M. Z. Bazant, and A. A. Kornyshev, J. Phys. Chem. B 125, 2677 (2021)
work page 2021
-
[53]
M. McEldrew, Z. A. Goodwin, S. Bi, A. A. Kornyshev, and M. Z. Bazant, J. Electrochem. Soc. 168, 050514 (2021)
work page 2021
-
[54]
M. McEldrew, Z. A. H. Goodwin, N. Molinari, B. Kozin- sky, A. A. Kornyshev, and M. Z. Bazant, J. Phys. Chem. B 125, 13752 (2021)
work page 2021
-
[55]
Z. A. H. Goodwin, M. McEldrew, J. de Souza, M. Z. Bazant, and A. A. Kornyshev, J. Chem. Phys. 157, 094106 (2022)
work page 2022
-
[56]
Z. A. H. Goodwin and A. A. Kornyshev, Electrochim. Acta 434, 141163 (2022)
work page 2022
-
[57]
P. J. Flory, J. Am. Chem. Soc. 63, 3083 (1941)
work page 1941
-
[58]
P. J. Flory, J. Am. Chem. Soc. 63, 3091 (1941)
work page 1941
-
[59]
P. J. Flory, J. Phys. Chem. 46, 132 (1942)
work page 1942
-
[60]
P. J. Flory, J. Chem. Phys. 10, 51 (1942)
work page 1942
-
[61]
P. J. Flory, Principles of polymer chemistry (Cornell Uni- versity Press, 1953)
work page 1953
- [62]
-
[63]
W. H. Stockmayer, J. Chem. Phys. 11, 45 (1943)
work page 1943
-
[64]
W. H. Stockmayer, J. Chem. Phys. 12, 125 (1944)
work page 1944
-
[65]
W. H. Stockmayer, J. Polym. Sci. 9, 69 (1952)
work page 1952
- [66]
- [67]
- [68]
- [69]
- [70]
- [71]
- [72]
- [73]
-
[74]
F. Tanaka, Polymer physics: applications to molecu- lar association and thermoreversible gelation (Cambridge University Press, 2011)
work page 2011
-
[75]
D. M. Markiewitz, Z. A. H. Goodwin, Q. Zheng, M. McEldrew, R. M. Espinosa-Marzal, and M. Z. Bazant, arXiv:2501.10578 (2025)
work page internal anchor Pith review Pith/arXiv arXiv 2025
- [76]
-
[77]
D. M. Markiewitz, Z. A. H. Goodwin, M. McEldrew, J. P. de Souza, X. Zhang, R. M. Espinosa-Marzal, and M. Z. Bazant, Faraday Discuss. 253, 365 (2024)
work page 2024
- [78]
- [79]
- [80]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.