Stability and Dynamics of Sn-based Halide Perovskites: Insights from MACE-MP-0 and Molecular Dynamics Simulations
Pith reviewed 2026-05-21 20:26 UTC · model grok-4.3
The pith
A general machine learning potential qualitatively predicts temperature-driven phase changes in tin halide perovskites
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
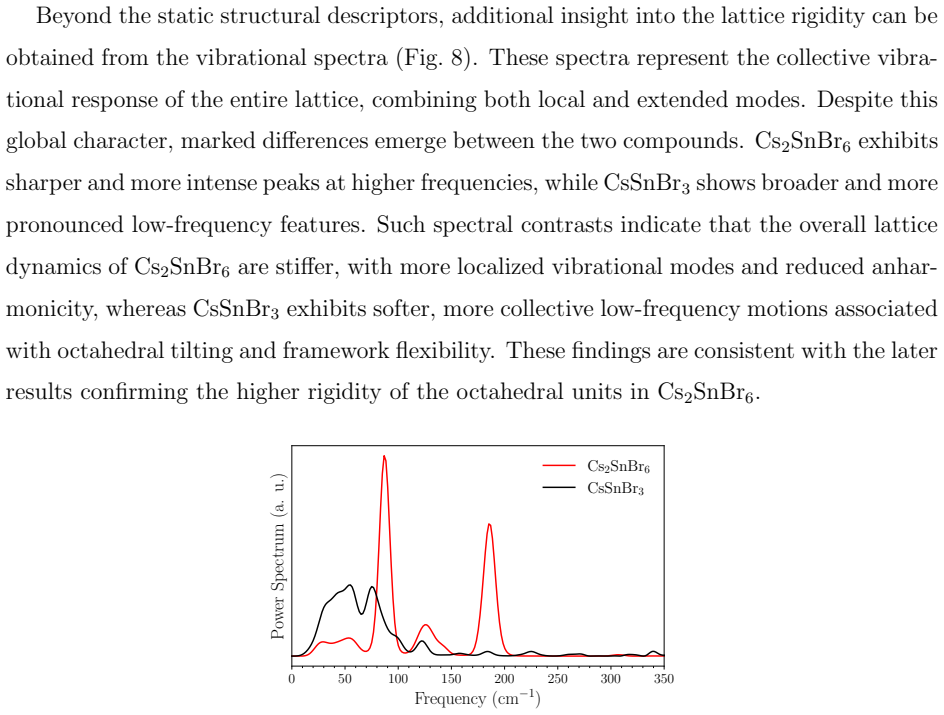
MACE-MP-0 applied without fine-tuning in NpT molecular dynamics simulations reproduces an orthorhombic-to-cubic phase transition in CsSnBr3, visible in lattice-parameter evolution and small anomalies in enthalpy and specific heat, although the intermediate tetragonal phase is not observed, while Cs2SnBr6 stays cubic with a more rigid octahedral framework across the full temperature range as indicated by radial distribution functions, bond-angle distributions, translational order parameters, and vibrational spectra.
What carries the argument
MACE-MP-0 interatomic potential driving NpT molecular dynamics simulations whose outputs are tracked through lattice parameters, enthalpy, specific heat, radial distribution functions, bond-angle distributions, and vibrational spectra
If this is right
- MACE-MP-0 can function as a practical first screening tool for thermal stability in other Sn-based halide perovskites.
- System-specific fine-tuning with density-functional-theory data is required when more subtle intermediate phases must be resolved.
- Multiple structural and thermodynamic descriptors together reliably distinguish rigid versus flexible octahedral frameworks.
- This workflow supports rapid qualitative assessment of how temperature affects lattice parameters and vibrational modes in these compounds.
Where Pith is reading between the lines
- Broadly trained potentials of this type may lower the barrier for initial computational exploration of many lead-free perovskite candidates.
- Applying the same protocol to mixed-halide or doped variants could map compositional trends in phase stability.
- Direct comparison of simulated specific-heat peaks against calorimetric measurements at several temperatures would quantify the model's limits on transition details.
Load-bearing premise
The broad training data of MACE-MP-0 transfers accurately enough to capture the temperature-driven phase behavior and octahedral framework rigidity in these specific Sn-halide perovskites without any fine-tuning or validation against DFT for the target systems.
What would settle it
If higher-accuracy DFT-based simulations or temperature-dependent diffraction experiments show that CsSnBr3 lacks an orthorhombic-to-cubic transition or exhibits markedly different specific-heat features between 100 K and 500 K, the claim of qualitative predictive power for MACE-MP-0 would be refuted.
Figures






read the original abstract
Tin-based halide perovskites have emerged as promising lead-free alternatives for optoelectronic applications, yet their structural stability and phase behavior at finite temperatures remain challenging to predict. Here, we assess the predictive capabilities of the foundational machine learning model MACE-MP-0 - trained on a broad chemical space and applied without system-specific fine-tuning - for the temperature-dependent behavior of CsSnBr3 and Cs2SnBr6. Molecular Dynamics simulations in the NpT ensemble were performed from 100 K to 500 K, and thermodynamic and structural descriptors including enthalpy, specific heat, radial distribution functions, translational order, bond angle distributions, and vibrational spectra were analyzed. Our results show that CsSnBr3 undergoes a low-temperature orthorhombic-to-cubic phase transition, evidenced by both the evolution of lattice parameters and subtle anomalies in enthalpy and specific heat, although the experimentally observed intermediate tetragonal phase is not captured. In contrast, Cs2SnBr6 remains cubic and maintains a more rigid octahedral framework across the entire temperature range. Overall, MACE-MP-0 qualitatively reproduces key thermal and structural features of these materials, highlighting its usefulness as a first step for studying new materials. For cases where capturing more subtle phase behavior is required, system-specific fine-tuning with Density Functional Theory data should be considered.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript evaluates the performance of the pre-trained MACE-MP-0 machine learning interatomic potential in molecular dynamics simulations of the tin-based halide perovskites CsSnBr3 and Cs2SnBr6. Using NpT ensemble MD from 100 to 500 K, the authors analyze thermodynamic properties (enthalpy, specific heat) and structural metrics (lattice parameters, radial distribution functions, bond angles, vibrational spectra) to assess phase stability and dynamics. They report that the model captures an orthorhombic-to-cubic transition in CsSnBr3 (though missing the experimental tetragonal phase) and the cubic stability with rigid octahedra in Cs2SnBr6, concluding that MACE-MP-0 is useful as a first step for studying such materials, with fine-tuning recommended for more subtle behaviors.
Significance. If the transferability of MACE-MP-0 holds without fine-tuning, this work would illustrate the utility of broad foundation models for rapid, qualitative screening of finite-temperature structural and thermodynamic properties in lead-free halide perovskites. The multi-descriptor analysis (enthalpy anomalies, RDFs, bond-angle distributions, and spectra) provides a coherent qualitative picture of octahedral rigidity and phase evolution, which is a strength for initial exploration in materials discovery.
major comments (3)
- [Abstract and Results] Abstract and Results sections: the central claim that MACE-MP-0 qualitatively reproduces the orthorhombic-to-cubic transition (via lattice parameters and enthalpy/specific-heat anomalies) in CsSnBr3 rests on unvalidated transferability; no system-specific DFT benchmarks for Sn-Br bonding, tilting energetics, or short MD runs are reported to anchor the observed features against model biases in the training distribution.
- [Methods] Methods section: the NpT MD protocol is conventional, but the manuscript provides no quantitative error bars, convergence tests with respect to system size or timestep, or direct experimental/DFT comparisons for key observables such as transition temperature or vibrational spectra, limiting verification of the reproduced phase behavior.
- [Results] Results section: while the missed tetragonal intermediate phase in CsSnBr3 is openly noted, there is no follow-up analysis (e.g., comparison of octahedral tilt angles or phonon modes) to diagnose whether this reflects a limitation in MACE-MP-0's description of anharmonicity for these specific compositions.
minor comments (2)
- [Figures] Figure captions and legends should explicitly state the temperature range and ensemble used for each plotted quantity to improve readability.
- [Methods] The definition of the translational order parameter and bond-angle distribution metrics should be given with explicit formulas in the Methods section rather than referenced only to prior work.
Simulated Author's Rebuttal
We thank the referee for their constructive and positive review, which recognizes the potential of MACE-MP-0 for qualitative exploration of lead-free perovskites. We address each major comment below and have revised the manuscript to strengthen the presentation of limitations and supporting analyses where feasible.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results sections: the central claim that MACE-MP-0 qualitatively reproduces the orthorhombic-to-cubic transition (via lattice parameters and enthalpy/specific-heat anomalies) in CsSnBr3 rests on unvalidated transferability; no system-specific DFT benchmarks for Sn-Br bonding, tilting energetics, or short MD runs are reported to anchor the observed features against model biases in the training distribution.
Authors: We agree that the absence of system-specific DFT benchmarks for Sn-Br interactions and tilting energetics limits the strength of the transferability claim. Our study deliberately examines zero-shot application of the pre-trained MACE-MP-0 model, with the transition supported by convergence across independent descriptors (lattice parameters, enthalpy anomalies, RDFs, and bond-angle distributions) that match experimental orthorhombic-to-cubic trends. In the revised manuscript we will add explicit discussion of this limitation and note that targeted DFT validation of tilting energetics would be a valuable follow-up for quantitative accuracy. revision: partial
-
Referee: [Methods] Methods section: the NpT MD protocol is conventional, but the manuscript provides no quantitative error bars, convergence tests with respect to system size or timestep, or direct experimental/DFT comparisons for key observables such as transition temperature or vibrational spectra, limiting verification of the reproduced phase behavior.
Authors: We accept that quantitative error bars, system-size convergence tests, and additional comparisons would improve verifiability. The revised manuscript will include error estimates on enthalpy and specific-heat curves, a supplementary note confirming convergence for the supercell sizes employed, and direct comparison of the computed vibrational spectra against available experimental Raman data for CsSnBr3. revision: yes
-
Referee: [Results] Results section: while the missed tetragonal intermediate phase in CsSnBr3 is openly noted, there is no follow-up analysis (e.g., comparison of octahedral tilt angles or phonon modes) to diagnose whether this reflects a limitation in MACE-MP-0's description of anharmonicity for these specific compositions.
Authors: We welcome the suggestion for diagnostic analysis. The revised Results section will incorporate temperature-dependent octahedral tilt-angle distributions and a brief comparison of low-frequency phonon modes to help identify whether the missing tetragonal phase arises from insufficient anharmonicity in the model for this composition. revision: yes
Circularity Check
No circularity: results from direct MD with external pre-trained model
full rationale
The paper applies the pre-trained MACE-MP-0 model (trained on broad Materials Project data) without fine-tuning or parameter adjustment to the target CsSnBr3/Cs2SnBr6 systems. Thermodynamic and structural descriptors (enthalpy, specific heat, lattice parameters, RDFs, bond angles, vibrational spectra) are computed directly from NpT MD trajectories at 100-500 K. These outputs emerge from the fixed interatomic potential and dynamics; no equations, fitted parameters, or self-citations define the target observables in terms of themselves. The qualitative reproduction of phase behavior is therefore an independent simulation result against external experimental benchmarks, not a reduction by construction.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption MACE-MP-0 provides sufficiently accurate forces for these Sn-based perovskites across 100-500 K without system-specific retraining.
- domain assumption NpT molecular dynamics with the chosen thermostat and barostat faithfully reproduces experimental thermal behavior.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
MACE-MP-0 foundational model … Molecular Dynamics simulations in the NpT ensemble … enthalpy, specific heat, radial distribution functions, translational order, bond angle distributions, and vibrational spectra
-
IndisputableMonolith/Foundation/DimensionForcing.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
CsSnBr3 undergoes a low-temperature orthorhombic-to-cubic phase transition … Cs2SnBr6 remains cubic and maintains a more rigid octahedral framework
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
- [1]
-
[2]
A. Inamuddin, A. Asiri, and K. Suvardhan,Green sustainable process for chemical and envi- ronmental engineering and science(Elsevier, 2019)
work page 2019
-
[3]
M. I. H. Ansari, A. Qurashi, and M. K. Nazeeruddin, Journal of Photochemistry and Photo- biology C: Photochemistry Reviews35, 1 (2018)
work page 2018
-
[4]
P. J. Dale and M. A. Scarpulla, Solar Energy Materials and Solar Cells251, 112097 (2023)
work page 2023
-
[5]
T. Li, X. Luo, P. Wang, Z. Li, Y. Li, J. Huang, Z. Jin, Y. Yang, B. Li, W. Zhang,et al., Nature , 1 (2025)
work page 2025
-
[6]
J. M. Frost and A. Walsh, Accounts of chemical research49, 528 (2016)
work page 2016
-
[7]
M. A. Carignano, S. A. Aravindh, I. S. Roqan, J. Even, and C. Katan, The Journal of Physical Chemistry C121, 20729 (2017)
work page 2017
-
[8]
R. Jinnouchi, J. Lahnsteiner, F. Karsai, G. Kresse, and M. Bokdam, Physical review letters 122, 225701 (2019). 16
work page 2019
- [9]
-
[10]
C. Yi, J. Luo, S. Meloni, A. Boziki, N. Ashari-Astani, C. Gr¨ atzel, S. M. Zakeeruddin, U. R¨ othlisberger, and M. Gr¨ atzel, Energy & Environmental Science9, 656 (2016)
work page 2016
-
[11]
R. E. Beal, D. J. Slotcavage, T. Leijtens, A. R. Bowring, R. A. Belisle, W. H. Nguyen, G. F. Burkhard, E. T. Hoke, and M. D. McGehee, The journal of physical chemistry letters7, 746 (2016)
work page 2016
- [12]
-
[13]
M. Ren, X. Qian, Y. Chen, T. Wang, and Y. Zhao, Journal of Hazardous Materials426, 127848 (2022)
work page 2022
-
[14]
S. F. Hoefler, G. Trimmel, and T. Rath, Monatshefte f¨ ur Chemie-Chemical Monthly148, 795 (2017)
work page 2017
-
[15]
A. K. Jena, A. Kulkarni, and T. Miyasaka, Chemical reviews119, 3036 (2019)
work page 2019
-
[16]
W. Ke, C. C. Stoumpos, and M. G. Kanatzidis, Advanced Materials31, 1803230 (2019)
work page 2019
- [17]
-
[18]
L. Lanzetta, N. Aristidou, and S. A. Haque, The Journal of Physical Chemistry Letters11, 574 (2020)
work page 2020
-
[19]
G. M. Dalpian, Q. Liu, C. C. Stoumpos, A. P. Douvalis, M. Balasubramanian, M. G. Kanatzidis, and A. Zunger, Physical Review Materials1, 025401 (2017)
work page 2017
- [20]
- [21]
-
[22]
P. Giannozzi, O. Baseggio, P. Bonf` a, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. De Gironcoli, P. Delugas, F. Ferrari Ruffino,et al., The Journal of chemical physics152 (2020)
work page 2020
-
[23]
T. D. K¨ uhne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Sch¨ utt, F. Schiffmann,et al., The Journal of Chemical Physics152(2020)
work page 2020
-
[24]
E. Fransson, J. M. Rahm, J. Wiktor, and P. Erhart, Chemistry of Materials35, 8229 (2023)
work page 2023
-
[25]
L. M. Farigliano, F. N. Ribeiro, and G. M. Dalpian, Materials Advances5, 5794 (2024)
work page 2024
-
[26]
X.-G. Zhao, G. M. Dalpian, Z. Wang, and A. Zunger, Physical Review B101, 155137 (2020). 17
work page 2020
- [27]
- [28]
-
[29]
E. Mosconi, J. M. Azpiroz, and F. De Angelis, Chemistry of materials27, 4885 (2015)
work page 2015
-
[30]
M. A. Carignano, A. Kachmar, and J. Hutter, The Journal of Physical Chemistry C119, 8991 (2015)
work page 2015
- [31]
-
[32]
A. P. Bart´ ok, M. C. Payne, R. Kondor, and G. Cs´ anyi, Physical review letters104, 136403 (2010)
work page 2010
-
[33]
A. P. Thompson, L. P. Swiler, C. R. Trott, S. M. Foiles, and G. J. Tucker, Journal of Com- putational Physics285, 316 (2015)
work page 2015
-
[34]
K. T. Sch¨ utt, H. E. Sauceda, P.-J. Kindermans, A. Tkatchenko, and K.-R. M¨ uller, The Journal of Chemical Physics148(2018)
work page 2018
-
[35]
V. L. Deringer, M. A. Caro, and G. Cs´ anyi, Advanced Materials31, 1902765 (2019)
work page 2019
-
[36]
S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, Nature communications13, 2453 (2022)
work page 2022
-
[37]
T. W. Ko and S. P. Ong, Nature Computational Science3, 998 (2023)
work page 2023
-
[38]
A foundation model for atomistic materials chemistry
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin,et al., arXiv preprint arXiv:2401.00096 (2023)
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[39]
I. Batatia, S. Batzner, D. P. Kov´ acs, A. Musaelian, G. N. Simm, R. Drautz, C. Ortner, B. Kozinsky, and G. Cs´ anyi, Nature Machine Intelligence , 1 (2025)
work page 2025
-
[40]
Drautz, Physical Review B99, 014104 (2019)
R. Drautz, Physical Review B99, 014104 (2019)
work page 2019
-
[41]
A. Jain, Y. Shin, and K. A. Persson, Nature Reviews Materials1, 1 (2016)
work page 2016
-
[42]
S. Hirotsu, J. Harada, M. Iizumi, and K. Gesi, Journal of the Physical Society of Japan37, 1393 (1974)
work page 1974
-
[43]
C. C. Stoumpos, C. D. Malliakas, J. A. Peters, Z. Liu, M. Sebastian, J. Im, T. C. Chasapis, A. C. Wibowo, D. Y. Chung, A. J. Freeman,et al., Crystal growth & design13, 2722 (2013)
work page 2013
-
[44]
N. Onoda-Yamamuro, T. Matsuo, and H. Suga, Journal of Physics and Chemistry of Solids 51, 1383 (1990). 18
work page 1990
-
[45]
P. Whitfield, N. Herron, W. Guise, K. Page, Y. Cheng, I. Milas, and M. Crawford, Scientific reports6, 35685 (2016)
work page 2016
-
[46]
C. Mao, X. He, H.-M. Lin, M. K. Gupta, P. Postec, T. Lanigan-Atkins, M. Krogstad, D. M. Pajerowski, T. Hong, T. J. Williams,et al., Physical Review Materials9, 065401 (2025)
work page 2025
- [47]
-
[48]
Plimpton, Journal of computational physics117, 1 (1995)
S. Plimpton, Journal of computational physics117, 1 (1995)
work page 1995
- [49]
-
[50]
J. R. Errington and P. G. Debenedetti, Nature409, 318 (2001)
work page 2001
-
[51]
M. Mori and H. Saito, Journal of Physics C: Solid State Physics19, 2391 (1986)
work page 1986
-
[52]
D. H. Fabini, G. Laurita, J. S. Bechtel, C. C. Stoumpos, H. A. Evans, A. G. Kontos, Y. S. Raptis, P. Falaras, A. Van der Ven, M. G. Kanatzidis,et al., Journal of the American Chemical Society138, 11820 (2016). 19
work page 2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.