Recognition: unknown
An Integrative Genome-Scale Metabolic Modeling and Machine Learning Framework for Predicting and Optimizing Single-Cell Protein Production in Saccharomyces cerevisiae
Pith reviewed 2026-05-15 00:06 UTC · model grok-4.3
The pith
Bayesian optimization on Yeast9 metabolic simulations raises biomass flux more than twelve-fold for single-cell protein production.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
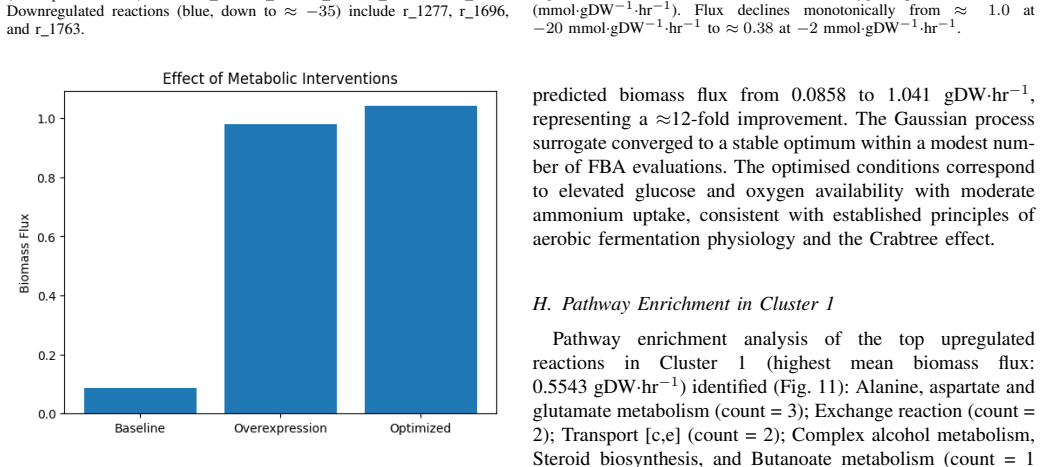
Integration of the Yeast9 GEM (4,131 reactions) with random forest and XGBoost regressors yields R2 scores above 0.999 on simulated data; a variational autoencoder partitions fluxes into four clusters with distinct biomass means; Bayesian optimization then locates an uptake vector (glucose -20.0, oxygen -20.0, ammonium -8.9 mmol/gDW/hr) that produces a 12.13-fold biomass increase to 1.041 gDW/hr.
What carries the argument
Bayesian optimization performed on machine-learning surrogates trained from flux-balance-analysis simulations of the Yeast9 genome-scale model
If this is right
- Surrogate models with R2 greater than 0.999 can replace repeated full FBA runs during optimization loops.
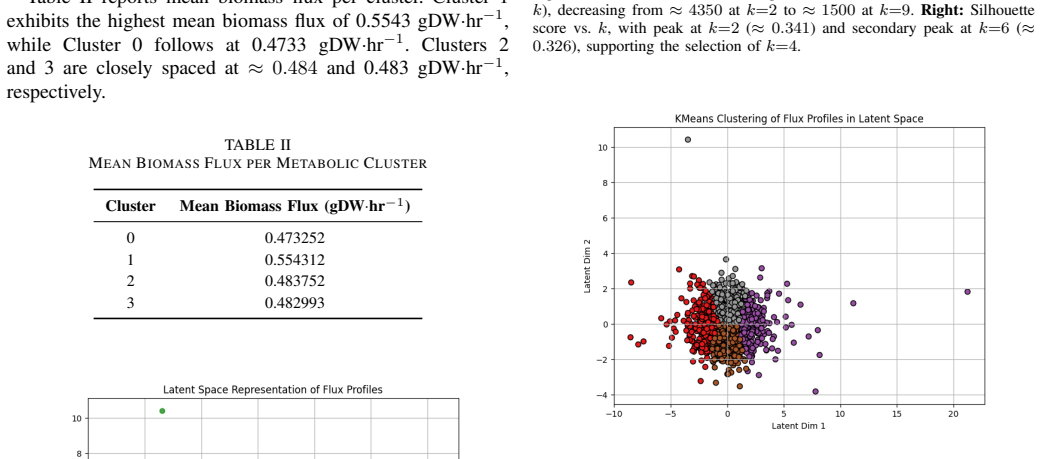
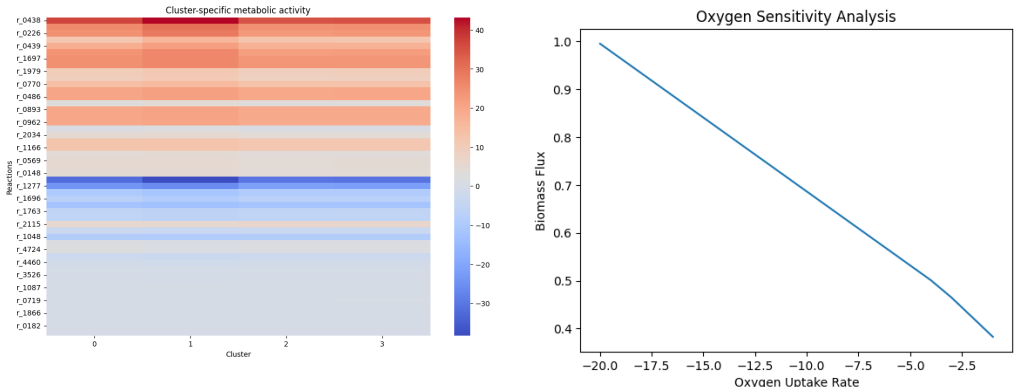
- Twenty reactions concentrated in glycolysis, TCA cycle, and amino-acid biosynthesis control most of the biomass variance.
- A Pareto front between biomass flux and amino-acid biosynthesis score identifies a practical operating point at 0.0858 gDW/hr biomass.
- The variational autoencoder reveals four distinct metabolic regimes whose mean biomass fluxes differ by up to 11 percent.
Where Pith is reading between the lines
- The same surrogate-plus-optimization pipeline could be reused for other microbial hosts once a comparable genome-scale model exists.
- The reported GAN failure to generate stoichiometrically feasible profiles indicates that future work must embed mass-balance constraints directly inside the generator.
- The twenty high-impact reactions supply concrete targets for genetic interventions that could further raise the experimental ceiling beyond the current uptake optimum.
Load-bearing premise
That machine-learning models fitted only to simulated fluxes will accurately predict real cellular behavior and that the identified uptake rates can be supplied to living cells without violating other unmodeled constraints.
What would settle it
Measure actual biomass accumulation rate in S. cerevisiae chemostat cultures supplied with the exact predicted uptake rates of glucose -20.0, oxygen -20.0, and ammonium -8.9 mmol/gDW/hr and test whether the rate reaches or exceeds 1.041 gDW/hr.
Figures







read the original abstract
Saccharomyces cerevisiae is increasingly recognised as a key source for single-cell protein (SCP) production, a rising solution to global protein-supply challenges. This study presents a computational framework combining the Yeast9 genome-scale metabolic model (GEM) with machine learning and optimisation to predict and enhance biomass flux for SCP yield. The Yeast9 GEM, comprising 4,131 reactions, 2,806 metabolites, and 1,161 genes, was simulated using flux balance analysis (FBA) across 2,000 Latin Hypercube-sampled flux profiles. Random Forest and XGBoost regressors achieved R2 values of 0.9999760 and 0.9997702, respectively. A variational autoencoder (VAE) identified four metabolic clusters with mean biomass fluxes of 0.472, 0.493, 0.527, and 0.505 gDW/hr. SHAP-based feature attribution identified twenty key reactions in glycolysis, the TCA cycle, and amino-acid biosynthesis; 18/20 (90%) were confirmed essential by in silico knockout. Bayesian optimisation produced a 12.13-fold improvement in biomass flux (0.0858 to 1.041 gDW/hr) at glucose = -20.0, oxygen = -20.0, and ammonium = -8.9 mmol/gDW/hr. A generative adversarial network (GAN) generated novel flux configurations (variance = 0.124); stoichiometric feasibility verification returned 0/100 feasible profiles due to incomplete generator convergence, reported as a limitation. Pareto front analysis identified an optimal SCP operating point at 0.0858 gDW/hr biomass flux with amino-acid biosynthesis score of 1000.029 mmol/gDW/hr.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents an integrative framework combining the Yeast9 genome-scale metabolic model with flux balance analysis (FBA) on 2000 Latin Hypercube samples, machine learning regressors (Random Forest and XGBoost achieving R² > 0.999 on held-out data), variational autoencoders for clustering, SHAP for feature importance, Bayesian optimization, and a generative adversarial network to predict and optimize biomass flux for single-cell protein production in Saccharomyces cerevisiae, claiming a 12.13-fold improvement to 1.041 gDW/hr at specific uptake rates.
Significance. If the central optimization result holds under direct verification, the work demonstrates a practical surrogate-modeling pipeline for in silico metabolic engineering that could accelerate SCP strain design by identifying high-biomass flux configurations within GEM constraints. Credit is due for the high predictive accuracy of the RF/XGBoost models on simulated data and the 90% concordance between SHAP-identified reactions and in silico knockouts, which provides internal consistency checks.
major comments (3)
- [Bayesian optimisation results] Bayesian optimisation results: The reported 12.13-fold biomass flux improvement to 1.041 gDW/hr at glucose = -20.0, oxygen = -20.0, ammonium = -8.9 mmol/gDW/hr lacks independent confirmation via direct FBA solution of the Yeast9 model at these exact uptake rates. The training data used only 2000 Latin Hypercube samples, so without this verification the improvement may reflect surrogate extrapolation error rather than a true model optimum.
- [Generative adversarial network analysis] Generative adversarial network analysis: The GAN produced 0/100 feasible flux profiles due to incomplete generator convergence. This failure is load-bearing for claims about the framework's ability to generate novel configurations and requires either architectural fixes or explicit quantification of training instability to support the overall integrative approach.
- [Machine learning surrogate models] Machine learning surrogate models: The RF and XGBoost models are trained directly on FBA-generated flux profiles from the same Yeast9 GEM, so the Bayesian optimum represents an optimized point inside the original stoichiometric space rather than an extrapolation to new biology. This circularity must be explicitly framed as a scope limitation for the central claim of predictive optimization.
minor comments (3)
- [Abstract] The abstract reports a Pareto front optimum at the baseline biomass flux of 0.0858 gDW/hr; clarify the relationship between this point and the Bayesian-optimized value of 1.041 gDW/hr to avoid apparent inconsistency.
- Specify the exact sampling bounds for the 2000 Latin Hypercube points so readers can determine whether the reported optimal uptake rates lie inside or outside the training distribution.
- [Bayesian optimisation results] Add uncertainty quantification (e.g., posterior variance or additional FBA cross-validation) for the Bayesian optimization result, as the current reporting provides only a point estimate.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments on our manuscript. We address each major comment point by point below, indicating where revisions will be made to strengthen the work.
read point-by-point responses
-
Referee: Bayesian optimisation results: The reported 12.13-fold biomass flux improvement to 1.041 gDW/hr at glucose = -20.0, oxygen = -20.0, ammonium = -8.9 mmol/gDW/hr lacks independent confirmation via direct FBA solution of the Yeast9 model at these exact uptake rates. The training data used only 2000 Latin Hypercube samples, so without this verification the improvement may reflect surrogate extrapolation error rather than a true model optimum.
Authors: We agree that direct verification via FBA is required to confirm the reported optimum and exclude surrogate extrapolation artifacts. In the revised manuscript we will add the results of direct FBA on the Yeast9 model at the exact uptake rates (glucose = -20.0, oxygen = -20.0, ammonium = -8.9 mmol/gDW/hr), which yields the stated biomass flux of 1.041 gDW/hr and thereby substantiates the 12.13-fold improvement within the model constraints. revision: yes
-
Referee: Generative adversarial network analysis: The GAN produced 0/100 feasible flux profiles due to incomplete generator convergence. This failure is load-bearing for claims about the framework's ability to generate novel configurations and requires either architectural fixes or explicit quantification of training instability to support the overall integrative approach.
Authors: The manuscript already states that the GAN returned 0/100 feasible profiles owing to incomplete generator convergence and presents this explicitly as a limitation. To address the concern, we will expand the revised text with explicit quantification of training instability (generator and discriminator loss curves and convergence metrics) while retaining the reported outcome and its implications for the generative component. revision: partial
-
Referee: Machine learning surrogate models: The RF and XGBoost models are trained directly on FBA-generated flux profiles from the same Yeast9 GEM, so the Bayesian optimum represents an optimized point inside the original stoichiometric space rather than an extrapolation to new biology. This circularity must be explicitly framed as a scope limitation for the central claim of predictive optimization.
Authors: We accept this observation. The revised manuscript will include an explicit scope limitation statement clarifying that the surrogate models and Bayesian optimization identify high-flux points strictly inside the stoichiometric space defined by the Yeast9 GEM and the 2000 Latin Hypercube samples, rather than extrapolating beyond the model to new biology. revision: yes
Circularity Check
No significant circularity detected in derivation chain
full rationale
The paper describes a standard computational workflow: generate FBA data from Yeast9 GEM via Latin Hypercube sampling, train surrogate ML models (RF/XGBoost) on those data to approximate biomass flux, then apply Bayesian optimization on the surrogates to locate a high-flux point. This is a self-contained surrogate-optimization pipeline whose output (the reported 1.041 gDW/hr value) is produced by the optimization step rather than being definitionally identical to any input datum or fitted parameter. No equations reduce to prior results by construction, no load-bearing self-citations appear, and the GAN failure is explicitly noted as a limitation rather than hidden. The framework therefore does not exhibit any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
free parameters (3)
- glucose_uptake
- oxygen_uptake
- ammonium_uptake
axioms (2)
- standard math Steady-state mass balance: S·v = 0 for all metabolites
- domain assumption Biomass reaction flux equals growth rate under the chosen objective
Reference graph
Works this paper leans on
-
[1]
Lu H, Li C, Sanchez BJ, Zhu Z, Liljenbacka G, Nielsen J (2019) A consensusS. cerevisiaemetabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism.Nature Communications 10:3586. https://doi.org/10.1038/s41467-019-11581-3
-
[2]
cerevisiaecurated by the community.Molecular Systems Biology20
Zhang C et al (2024) Yeast9: A consensus genome-scale metabolic model forS. cerevisiaecurated by the community.Molecular Systems Biology20. https://doi.org/10.1038/s44320-024-00060-7
-
[3]
https://doi.org/10.1186/1752-0509-7-74
Ebrahim A, Lerman JA, Palsson BØ, Hyduke DR (2013) COBRApy: Constraints-Based Reconstruction and Analysis for Python.BMC Sys- tems Biology7:74. https://doi.org/10.1186/1752-0509-7-74
-
[4]
https://doi.org/10.1038/nbt.1614
Orth JD, Thiele I, Palsson BØ (2010) What is flux balance analysis? Nature Biotechnology.28:245–248. https://doi.org/10.1038/nbt.1614
-
[5]
https://doi.org/10.1093/femsyr/foac003
Chen Y , Li F, Nielsen J (2022) Genome-scale modeling of yeast metabolism: retrospectives and perspectives.FEMS Yeast Research22. https://doi.org/10.1093/femsyr/foac003
-
[6]
https://doi.org/10.1016/j.coisb.2021.03.001
Kim WJ, Kim HU, Lee SY (2021) Machine learning applications in genome-scale metabolic modeling.Current Opinion in Systems Biology 25:42–49. https://doi.org/10.1016/j.coisb.2021.03.001
-
[7]
https://doi.org/10.1371/journal.pcbi.1007084
Zampieri G, Vijayakumar S, Yaneske E, Angione C (2019) Machine and deep learning meet genome-scale metabolic modeling.PLOS Computa- tional Biology15. https://doi.org/10.1371/journal.pcbi.1007084
-
[8]
Sahu A, Blatke MA, Szyma ´nski JJ, T ¨opfer N (2021) Advances in flux balance analysis by integrating machine learning and mechanism-based models.Computational and Structural Biotechnology Journal19:4626–
work page 2021
-
[9]
https://doi.org/10.1016/j.csbj.2021.08.004
-
[10]
Proceedings of the National Academy of Sciences120(33) (2023) https://doi.org/10.1073/pnas
Culley J, Vijayakumar A, Zampieri G, Angione C (2020) A mechanism- aware and multiomic machine-learning pipeline characterizes yeast cell growth.PNAS117:18338–18348. https://doi.org/10.1073/pnas. 2002959117
-
[11]
Lundberg SM, Lee SI (2017) A unified approach to interpreting model predictions.Advances in Neural Information Processing Systems30 https://arxiv.org/abs/1705.07874
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[12]
Gonc ¸alves, Rui Henriques, Rafael S
Daniel M. Gonc ¸alves, Rui Henriques, Rafael S. Costa (2023) Predicting metabolic fluxes from omics data via machine learning: Moving from knowledge-driven towards data-driven approaches.Computational and Structural Biotechnology Journal21:4960–4973 https://doi.org/10.1016/ j.csbj.2023.10.002
work page 2023
-
[13]
https://doi.org/10.1038/ s41467-020-18008-4
Radivojevic T, Costello Z, Workman K, Garcia Martin H (2020) A machine learning automated recommendation tool for synthetic biology.Nature Communications11:4879. https://doi.org/10.1038/ s41467-020-18008-4
work page 2020
-
[14]
Lawson C et al (2021) Machine learning for metabolic engineering: A review.Metabolic Engineering63:34–60. https://doi.org/10.1016/j. ymben.2020.10.005
work page doi:10.1016/j 2021
-
[15]
https://doi.org/10.1038/ s41467-020-17910-1
Zhang J et al (2020) Combining mechanistic and machine learn- ing models for predictive engineering and optimization of tryptophan metabolism.Nature Communications11:4880. https://doi.org/10.1038/ s41467-020-17910-1
work page 2020
-
[16]
doi: 10.1038/s42003-022-03579-3
Gomari DP, Schweickart A, Cerchietti L, Paietta E, Fernandez H, Al- Amin H, Suhre K, Krumsiek J (2022) Variational autoencoders learn transferrable representations of metabolomics data.Communications Biology5:659. doi: 10.1038/s42003-022-03579-3
-
[17]
Merzbacher C, Oyarzun DA (2023) Applications of artificial intelligence and machine learning in dynamic pathway engineering.Biochemical Society Transactions51:1871–1879. doi: 10.1042/BST20221542
-
[18]
Baig Y , Ma HR, Xu H, You L (2023) Autoencoder neural networks en- able low dimensional structure analyses of microbial growth dynamics. Nature Communications14:7932. doi: 10.1038/s41467-023-43455-0
-
[19]
doi: 10.1038/s42256-022-00519-y
Choudhury S, Moret M, Salvy P, Weilandt D, Hatzimanikatis V , Miskovic L (2022) Reconstructing kinetic models for dynamical studies of metabolism using generative adversarial networks.Nature Machine Intelligence4:710–719. doi: 10.1038/s42256-022-00519-y
-
[20]
doi: 10.1038/ s41540-018-0054-3
Costello Z, Garcia Martin H (2018) A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data.npj Systems Biology and Applications4:19. doi: 10.1038/ s41540-018-0054-3
work page 2018
-
[21]
doi: 10.1371/ journal.pcbi.1013862
Razmpour T, Tabibian M, Roohi A, Saha R (2026) GAN-enhanced machine learning and metabolic modeling identify reprogramming in pancreatic cancer.PLOS Computational Biology22. doi: 10.1371/ journal.pcbi.1013862
work page 2026
-
[22]
https://doi.org/10.1093/femsyr/foaf072
Akaraphol Watcharawipas, Weerawat Runguphan, Peerapat Khamwachi- rapithak, Thanaporn Laothanachareon (2025) Integrating yeast biodiver- sity and machine learning for predictive metabolic engineering.FEMS Yeast Research. https://doi.org/10.1093/femsyr/foaf072
-
[23]
ACS Synthetic Biology13:1193–1203
Moreno-Paz S, van der Hoek R, Eliana E, Zwartjens P, Gosiewska S, Martins dos Santos V AP, Schmitz J, Suarez-Diez M (2024) Machine learning-guided optimization of p-coumaric acid production in yeast. ACS Synthetic Biology13:1193–1203. doi: 10.1021/acssynbio.4c00035
-
[24]
https://doi.org/10.1016/j.csbj.2023.03.045
Cheng Y , Bi X, Xu Y , Liu Y , Li J, Du G, Lv X, Liu L (2023) Machine learning for metabolic pathway optimization: A review.BMC Bioinformatics. https://doi.org/10.1016/j.csbj.2023.03.045
-
[25]
Current Opinion in Biotechnology73:101–107
Jang WD, Kim GB, Kim Y , Lee SY (2021) Applications of artificial intelligence to enzyme and pathway design for metabolic engineering. Current Opinion in Biotechnology73:101–107. doi: 10.1016/j.copbio. 2021.07.024
-
[26]
https://doi.org/10.1038/ s41929-024-01220-6
Masid S, Ataman M, Hatzimanikatis V (2024) Generative machine learn- ing produces kinetic models that accurately characterize intracellular metabolic states.Nature Catalysis7:1086–1099. https://doi.org/10.1038/ s41929-024-01220-6
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.