Recognition: no theorem link
Does the total energy difference method for modelling core level photoemission fail for bigger molecules?
Pith reviewed 2026-05-10 19:05 UTC · model grok-4.3
The pith
New experimental data for anthrone shows the total energy difference method accurately predicts core electron binding energies even in larger molecules.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the Δ-self-consistent-field method does not lose accuracy for larger molecules. Fresh gas-phase measurements of anthrone's C 1s binding energies differ markedly from prior literature but agree well with ΔSCF results obtained with the SCAN functional. When the same approach is applied to 44 core binding energies across molecules containing 10 to 40 atoms, the mean absolute error is 0.19 eV, matching the performance seen on small-molecule benchmarks. These findings, together with general theoretical considerations, indicate that the total energy difference method is suitable for localized excitations in both small and large molecules.
What carries the argument
The Δ-self-consistent-field (ΔSCF) method, which obtains core electron binding energies as the difference in total energy between the neutral ground state and a core-hole state computed self-consistently.
If this is right
- The total energy difference method can be applied reliably to core level calculations on molecules containing between 10 and 40 atoms.
- Earlier large errors reported for anthrone originated in inaccuracies in the published experimental spectrum rather than in the computational approach.
- The method's performance on the 44-value dataset is consistent with prior small-molecule benchmarks, supporting its use for localized excitations without size-dependent degradation.
- General theoretical considerations in the paper suggest the approach remains promising for other extended systems.
Where Pith is reading between the lines
- If the agreement generalizes, routine computational modeling of core photoemission in larger organic and biological molecules can rely on this efficient total-energy-difference route rather than switching to more costly many-body methods.
- Re-examination of experimental spectra for other molecules where the method previously appeared to fail could resolve similar discrepancies.
- The choice of a functional that handles core-hole states well, such as SCAN, may be sufficient to maintain accuracy without empirical per-molecule tuning.
Load-bearing premise
The newly recorded gas-phase photoelectron spectrum of anthrone is free of systematic calibration errors or sample impurities, and the SCAN functional does not require molecule-by-molecule adjustments to produce the reported agreement.
What would settle it
An independent gas-phase C 1s photoelectron spectrum of anthrone that reproduces the older published binding energies instead of the new values reported in this work.
Figures

read the original abstract
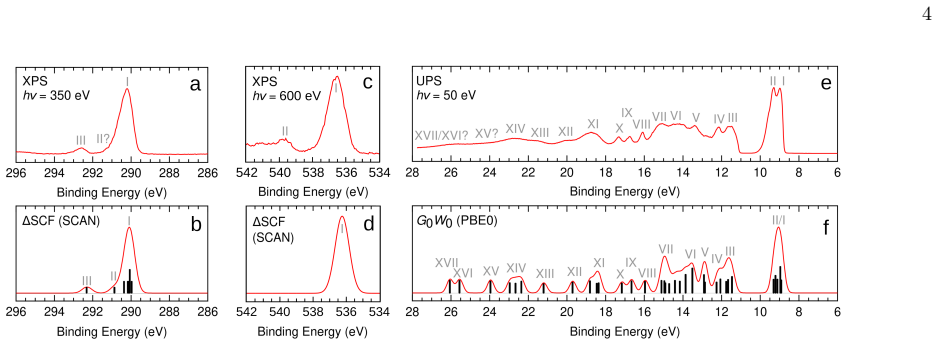
The $\Delta$-Self-Consistent-Field ($\Delta$SCF) method permits calculations of core electron binding energies in materials and molecules at a modest computational cost. However, it has been reported that whilst this method works well for small molecules, its accuracy drops off dramatically when larger systems are considered. Particularly large errors have been reported for the anthrone molecule, which consists of 25 atoms. In this work, the gas-phase photoelectron spectrum of anthrone is revisited both computationally and experimentally. The measured C 1s binding energies in anthrone differ markedly from previously published values, and the new experimental results are in good agreement with $\Delta$SCF calculations based on the SCAN functional. In addition, the performance of the $\Delta$SCF method is evaluated for a dataset of 44 core electron binding energies from medium sized molecules containing between 10 and 40 atoms. The mean absolute error for this dataset - 0.19 eV - is comparable to the results of previous computational benchmarks. Overall, these results and general theoretical considerations indicate that the $\Delta$SCF method is suitable for modelling localized excitations in both small and large molecules, and applications to other extended systems are also promising.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that the ΔSCF method for core electron binding energies does not fail for larger molecules. This is supported by new experimental gas-phase C 1s photoelectron data for anthrone (25 atoms) that differ from previous literature values and agree with ΔSCF calculations using the SCAN meta-GGA functional. The authors also report a mean absolute error of 0.19 eV for ΔSCF on a dataset of 44 core binding energies from molecules ranging from 10 to 40 atoms, which is comparable to benchmarks for smaller systems. They conclude that the method is suitable for localized excitations in both small and large molecules.
Significance. If the new experimental results hold, the work is significant as it addresses and potentially resolves reported failures of ΔSCF for medium-sized molecules like anthrone, providing evidence that the method remains accurate with a modest computational cost. The compilation of 44 binding energies and the low MAE offer a useful benchmark for the community. The paper credits the agreement with SCAN-ΔSCF without parameter fitting, which is a strength. This could encourage broader application to extended systems where more expensive methods are prohibitive.
major comments (1)
- The revised C 1s binding energies for anthrone are pivotal to the paper's central claim, as they 'differ markedly' from prior values and align with theory (abstract). However, the manuscript lacks specific details on the absolute calibration procedure (e.g., reference lines, simultaneous standards, or work function), sample purity verification (e.g., mass spectrometry or temperature checks during sublimation), and the deconvolution method for the overlapping C 1s peaks. Without these, systematic errors cannot be ruled out as the source of the discrepancy with earlier experiments.
minor comments (2)
- The title poses a question; a declarative title might better reflect the affirmative conclusion reached in the abstract and discussion.
- The abstract refers to 'general theoretical considerations' supporting the conclusion; these should be briefly outlined or referenced in the main text for completeness.
Simulated Author's Rebuttal
We thank the referee for their positive evaluation of the significance of our work and for the recommendation of major revision. We address the single major comment below in detail.
read point-by-point responses
-
Referee: The revised C 1s binding energies for anthrone are pivotal to the paper's central claim, as they 'differ markedly' from prior values and align with theory (abstract). However, the manuscript lacks specific details on the absolute calibration procedure (e.g., reference lines, simultaneous standards, or work function), sample purity verification (e.g., mass spectrometry or temperature checks during sublimation), and the deconvolution method for the overlapping C 1s peaks. Without these, systematic errors cannot be ruled out as the source of the discrepancy with earlier experiments.
Authors: We agree that the experimental details are essential for validating the new anthrone C 1s binding energies and for allowing independent assessment of possible systematic differences with prior literature. In the revised manuscript we will expand the experimental methods section to provide: (i) the full absolute calibration procedure, including the specific reference lines, any simultaneous standards, and work-function considerations; (ii) sample-purity verification via mass spectrometry together with the temperature monitoring protocol used during sublimation; and (iii) a complete description of the peak-deconvolution procedure for the overlapping C 1s features, specifying the fitting functions, constraints, and software employed. These additions will directly address the referee’s concern and strengthen the central claim. revision: yes
Circularity Check
No circularity: direct empirical comparison of new measurements to standard ΔSCF
full rationale
The paper reports new gas-phase C 1s photoelectron spectra for anthrone, notes that these differ from prior literature values, and shows agreement with unmodified ΔSCF calculations using the SCAN functional. It further benchmarks the same method on an independent set of 44 core-level binding energies from 10–40 atom molecules, obtaining an MAE of 0.19 eV. No parameters are fitted to the target observables, no self-referential definitions or equations are introduced, and the central claim rests on external experimental data rather than self-citations, ansatzes, or uniqueness theorems imported from the authors’ prior work. The derivation chain is therefore self-contained as a straightforward validation exercise.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
In the implementation used in this work [43], the self-energy is first calculated on the imaginary frequency axis, and then analytically continued onto the real axis
starting point. In the implementation used in this work [43], the self-energy is first calculated on the imaginary frequency axis, and then analytically continued onto the real axis. For the analytic continuation, a Pade approximant with 16 parameters was employed. Quadruple-zeta numerical atom-centered basis sets with valence-correlation consistency from...
-
[2]
The measured peak positions are significantly different from the values reported in ref
The experimental C 1s spectrum (Figure 2a) contains two main features: a strong, broad, and asymmetric peak at 290.27 eV, and a weaker peak at 292.7 eV. The measured peak positions are significantly different from the values reported in ref. [57, 58]: 290.72 eV and 293.6 eV, respectively (see the comparison in Table I). Due to this discrepancy, the method...
-
[3]
Diptarka Hait and Martin Head-Gordon. Excited State Orbital Optimization via Minimizing the Square of the Gradient: General Approach and Application to Singly and Doubly Excited States via Density Functional Theory.J. Chem. Theory Comput., 16(3): 1699–1710, March 2020. ISSN 1549-9618, 1549-9626. doi: 10.1021/acs.jctc.9b01127
-
[4]
Orbital Optimized Density Functional Theory for Electronic Excited States.J
Diptarka Hait and Martin Head-Gordon. Orbital Optimized Density Functional Theory for Electronic Excited States.J. Phys. Chem. Lett., 12(19):4517– 4529, May 2021. ISSN 1948-7185, 1948-7185. doi: 10.1021/acs.jpclett.1c00744
-
[5]
Matthias Kahk and Johannes Lischner
J. Matthias Kahk and Johannes Lischner. Combining the ∆-Self-Consistent-Field and GW Methods for Predicting Core Electron Binding Energies in Periodic Solids.J. Chem. Theory Comput., 19(11):3276– 3283, June 2023. ISSN 1549-9618, 1549-9626. doi: 10.1021/acs.jctc.3c00121
-
[6]
Samuel J. Hall, Benedikt P. Klein, and Reinhard J. Maurer. Characterizing Molecule–Metal Surface Chemistry with Ab Initio Simulation of X-ray Absorption and Photoemission Spectra.J. Phys. Chem. C, 127(4): 1870–1880, February 2023. ISSN 1932-7447, 1932-7455. doi:10.1021/acs.jpcc.2c06996
-
[7]
Misa Nishino, Kouji Inagaki, Yoshitada Morikawa, Kazuya Yamamura, and Yuji Ohkubo. Identification of chemical species on plasma-treated polytetrafluoroethylene surface by ab-initio calculations of core-energy-level shift in X-ray photoelectron spectra. Applied Surface Science, 655:159369, May 2024. ISSN 01694332. doi:10.1016/j.apsusc.2024.159369
-
[8]
Kevin Carter-Fenk and John M. Herbert. State-Targeted Energy Projection: A Simple and Robust Approach to Orbital Relaxation of Non-Aufbau Self-Consistent Field Solutions.J. Chem. Theory Comput., 16(8):5067– 5082, August 2020. ISSN 1549-9618, 1549-9626. doi: 7 10.1021/acs.jctc.0c00502
-
[9]
Excited state dipole moments from ∆SCF: a benchmark.Phys
Lukas Paetow and Johannes Neugebauer. Excited state dipole moments from ∆SCF: a benchmark.Phys. Chem. Chem. Phys., 27(31):16354–16370, 2025. ISSN 1463- 9076, 1463-9084. doi:10.1039/D5CP01695A
-
[10]
Ethan Pollack, Rohan Maniar, and John P. Perdew. ∆SCF Excitation Energies up a Ladder of Ground-State Density Functionals.J. Phys. Chem. A, 129(47):11014– 11020, November 2025. ISSN 1089-5639, 1520-5215. doi: 10.1021/acs.jpca.5c05336
-
[11]
P. S. Bagus. Self-Consistent-Field Wave Functions for Hole States of Some Ne-Like and Ar-Like Ions.Phys. Rev., 139(3A):A619–A634, August 1965. ISSN 0031- 899X. doi:10.1103/PhysRev.139.A619
-
[12]
M.S. Banna, R.J. Key, and C.S. Ewig. Accurate core binding energies of ions from Dirac-Fock calculations combined with experimental atomic binding energies.Journal of Electron Spectroscopy and Related Phenomena, 26(3):259–266, January 1982. ISSN 03682048. doi:10.1016/0368-2048(82)85033-0
-
[13]
L. Triguero, O. Plashkevych, L.G.M. Pettersson, and H. ˚Agren. Separate state vs. transition state Kohn-Sham calculations of X-ray photoelectron binding energies and chemical shifts.Journal of Electron Spectroscopy and Related Phenomena, 104(1-3):195–207, July 1999. ISSN 03682048. doi:10.1016/S0368-2048(99)00008-0
-
[14]
Nicholas A. Besley, Andrew T. B. Gilbert, and Peter M. W. Gill. Self-consistent-field calculations of core excited states.The Journal of Chemical Physics, 130 (12):124308, March 2009. ISSN 0021-9606, 1089-7690. doi:10.1063/1.3092928
-
[15]
Bagus, Eugene Ilton, and Connie J
Paul S. Bagus, Eugene Ilton, and Connie J. Nelin. Extracting Chemical Information from XPS Spectra: A Perspective.Catal Lett, 148(7):1785–1802, July 2018. ISSN 1011-372X, 1572-879X. doi:10.1007/s10562-018- 2417-1
-
[16]
Matthias Kahk and Johannes Lischner
J. Matthias Kahk and Johannes Lischner. Accurate absolute core-electron binding energies of molecules, solids, and surfaces from first-principles calculations. Phys. Rev. Materials, 3(10):100801, October 2019. ISSN 2475-9953. doi:10.1103/PhysRevMaterials.3.100801
-
[17]
Diptarka Hait and Martin Head-Gordon. Highly Accurate Prediction of Core Spectra of Molecules at Density Functional Theory Cost: Attaining Sub- electronvolt Error from a Restricted Open-Shell Kohn–Sham Approach.J. Phys. Chem. Lett., 11(3): 775–786, February 2020. ISSN 1948-7185, 1948-7185. doi:10.1021/acs.jpclett.9b03661
-
[18]
J. Matthias Kahk, Georg S. Michelitsch, Reinhard J. Maurer, Karsten Reuter, and Johannes Lischner. Core Electron Binding Energies in Solids from Periodic All- Electron ∆-Self-Consistent-Field Calculations.J. Phys. Chem. Lett., 12(38):9353–9359, September 2021. ISSN 1948-7185, 1948-7185. doi:10.1021/acs.jpclett.1c02380
-
[19]
Benedikt P Klein, Samuel J Hall, and Reinhard J Maurer. The nuts and bolts of core-hole constrained ab initio simulation for K-shell x-ray photoemission and absorption spectra.J. Phys.: Condens. Matter, 33(15): 154005, April 2021. ISSN 0953-8984, 1361-648X. doi: 10.1088/1361-648X/abdf00
-
[20]
Nicholas A. Besley. Modeling of the spectroscopy of core electrons with density functional theory.WIREs Comput Mol Sci, 11(6):e1527, November 2021. ISSN 1759-0876, 1759-0884. doi:10.1002/wcms.1527
-
[21]
Weitao Yang and Paul W. Ayers. Foundation for the ∆SCF Approach in Density Functional Theory, March
- [22]
-
[23]
Performance of the TPSS Functional on Predicting Core Level Binding Energies of Main Group Elements Containing Molecules: A Good Choice for Molecules Adsorbed on Metal Surfaces.J
No` elia Pueyo Bellafont, Francesc Vi˜ nes, and Francesc Illas. Performance of the TPSS Functional on Predicting Core Level Binding Energies of Main Group Elements Containing Molecules: A Good Choice for Molecules Adsorbed on Metal Surfaces.J. Chem. Theory Comput., 12(1):324–331, January 2016. ISSN 1549-9618, 1549-
2016
-
[24]
doi:10.1021/acs.jctc.5b00998
-
[25]
Haugen, Zheyue Yang, Katherine J
Diptarka Hait, Eric A. Haugen, Zheyue Yang, Katherine J. Oosterbaan, Stephen R. Leone, and Martin Head-Gordon. Accurate prediction of core-level spectra of radicals at density functional theory cost via square gradient minimization and recoupling of mixed configurations.The Journal of Chemical Physics, 153 (13):134108, October 2020. ISSN 0021-9606, 1089-7...
-
[26]
Caldas, Patrick Rinke, Volker Blum, and Matthias Scheffler
Max Pinheiro, Marilia J. Caldas, Patrick Rinke, Volker Blum, and Matthias Scheffler. Length dependence of ionization potentials of transacetylenes: Internally consistent DFT/ G W approach.Phys. Rev. B, 92(19): 195134, November 2015. ISSN 1098-0121, 1550-235X. doi:10.1103/PhysRevB.92.195134
-
[27]
Fabiano Corsetti and Arash A. Mostofi. System- size convergence of point defect properties: The case of the silicon vacancy.Phys. Rev. B, 84(3): 035209, July 2011. ISSN 1098-0121, 1550-235X. doi: 10.1103/PhysRevB.84.035209
-
[28]
n -type doping of CuIn Se 2 and CuGa Se 2
Clas Persson, Yu-Jun Zhao, Stephan Lany, and Alex Zunger. n -type doping of CuIn Se 2 and CuGa Se 2. Phys. Rev. B, 72(3):035211, July 2005. ISSN 1098-0121, 1550-235X. doi:10.1103/PhysRevB.72.035211
-
[29]
Dorothea Golze, Jan Wilhelm, Michiel J. Van Setten, and Patrick Rinke. Core-Level Binding Energies from GW: An Efficient Full-Frequency Approach within a Localized Basis.J. Chem. Theory Comput., 14(9):4856– 4869, September 2018. ISSN 1549-9618, 1549-9626. doi: 10.1021/acs.jctc.8b00458
-
[30]
Accurate Absolute and Relative Core-Level Binding Energies fromGW.J
Dorothea Golze, Levi Keller, and Patrick Rinke. Accurate Absolute and Relative Core-Level Binding Energies fromGW.J. Phys. Chem. Lett., 11(5):1840– 1847, March 2020. ISSN 1948-7185, 1948-7185. doi: 10.1021/acs.jpclett.9b03423
-
[31]
Dorothea Golze, Markus Hirvensalo, Patricia Hern´ andez-Le´ on, Anja Aarva, Jarkko Etula, Toma Susi, Patrick Rinke, Tomi Laurila, and Miguel A. Caro. Accurate Computational Prediction of Core- Electron Binding Energies in Carbon-Based Materials: A Machine-Learning Model Combining Density- Functional Theory andGW.Chem. Mater., 34(14): 6240–6254, July 2022....
-
[32]
Benchmark of$\boldsymbol{GW}$ Methods for Core-Level Binding Energies, 2022
Jiachen Li, Ye Jin, Patrick Rinke, Weitao Yang, and Dorothea Golze. Benchmark of$\boldsymbol{GW}$ Methods for Core-Level Binding Energies, 2022. Version Number: 1
2022
-
[33]
Iskander Mukatayev, Benoˆ ıt Skl´ enard, Valerio Olevano, and Jing Li. Electron removal energies in noble- gas atoms up to 100 keV:Ab initioG W versus x- ray photoelectron spectroscopy.Phys. Rev. B, 106(8): L081125, August 2022. ISSN 2469-9950, 2469-9969. doi: 10.1103/PhysRevB.106.L081125. 8
-
[34]
Matthias Kahk and Johannes Lischner
J. Matthias Kahk and Johannes Lischner. Predicting core electron binding energies in elements of the first transition series using the ∆-self-consistent-field method. Faraday Discuss., 236:364–373, 2022. ISSN 1359-6640, 1364-5498. doi:10.1039/D1FD00103E
-
[35]
Volker Blum, Ralf Gehrke, Felix Hanke, Paula Havu, Ville Havu, Xinguo Ren, Karsten Reuter, and Matthias Scheffler. Ab initio molecular simulations with numeric atom-centered orbitals.Computer Physics Communications, 180(11):2175–2196, November 2009. ISSN 00104655. doi:10.1016/j.cpc.2009.06.022
-
[36]
Joseph W. Abbott, Carlos Mera Acosta, Alaa Akkoush, Alberto Ambrosetti, Viktor Atalla, Alexej Bagrets, J¨ org Behler, Daniel Berger, Bj¨ orn Bieniek, Jonas Bj¨ ork, Volker Blum, Saeed Bohloul, Connor L. Box, Nicholas Boyer, Danilo Simoes Brambila, Gabriel A. Bramley, Kyle R. Bryenton, Mar´ ıa Camarasa-G´ omez, Christian Carbogno, Fabio Caruso, Sucismita C...
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[37]
Victor Wen-zhe Yu, Fabiano Corsetti, Alberto Garc´ ıa, William P. Huhn, Mathias Jacquelin, Weile Jia, Bj¨ orn Lange, Lin Lin, Jianfeng Lu, Wenhui Mi, Ali Seifitokaldani, ´Alvaro V´ azquez-Mayagoitia, Chao Yang, Haizhao Yang, and Volker Blum. ELSI: A unified software interface for Kohn–Sham electronic structure solvers.Computer Physics Communications, 222:...
-
[38]
V. Havu, V. Blum, P. Havu, and M. Scheffler. Efficient integration for all-electron electronic structure calculation using numeric basis functions.Journal of Computational Physics, 228(22):8367–8379, December
-
[39]
ISSN 00219991. doi:10.1016/j.jcp.2009.08.008
-
[40]
Jianwei Sun, Adrienn Ruzsinszky, and John P. Perdew. Strongly Constrained and Appropriately Normed Semilocal Density Functional.Phys. Rev. Lett., 115(3):036402, July 2015. ISSN 0031-9007, 1079-7114. doi:10.1103/PhysRevLett.115.036402
-
[41]
R. Strange, F.R. Manby, and P.J. Knowles. Automatic code generation in density functional theory.Computer Physics Communications, 136(3):310–318, May 2001. ISSN 00104655. doi:10.1016/S0010-4655(01)00148-5
-
[42]
Jorge Nocedal and Stephen J. Wright.Numerical Optimization. Springer Series in Operations Research and Financial Engineering. Springer New York, 2006. ISBN 978-0-387-30303-1. doi:10.1007/978-0-387-40065-5
-
[43]
C. G. Broyden. The Convergence of a Class of Double-rank Minimization Algorithms 1. General Considerations.IMA J Appl Math, 6(1):76–90, 1970. ISSN 0272-4960, 1464-3634. doi:10.1093/imamat/6.1.76
-
[44]
R. Fletcher. A new approach to variable metric algorithms.The Computer Journal, 13(3):317–322, March 1970. ISSN 0010-4620, 1460-2067. doi: 10.1093/comjnl/13.3.317
-
[46]
D. F. Shanno. Conditioning of quasi-Newton methods for function minimization.Math. Comp., 24(111):647–656,
-
[47]
doi:10.1090/S0025- 5718-1970-0274029-X
ISSN 0025-5718, 1088-6842. doi:10.1090/S0025- 5718-1970-0274029-X
-
[48]
The Journal of Chemical Physics , author =
Carlo Adamo and Vincenzo Barone. Toward reliable density functional methods without adjustable parameters: The PBE0 model.The Journal of Chemical Physics, 110(13):6158–6170, April 1999. ISSN 0021-9606, 1089-7690. doi:10.1063/1.478522
-
[50]
Tailoring of arbitrary optical vector beams
Igor Ying Zhang, Xinguo Ren, Patrick Rinke, Volker Blum, and Matthias Scheffler. Numeric atom-centered- orbital basis sets with valence-correlation consistency from H to Ar.New J. Phys., 15(12):123033, December 2013. ISSN 1367-2630. doi:10.1088/1367- 2630/15/12/123033
-
[51]
U. Gelius and K. Siegbahn. ESCA studies of molecular core and valence levels in the gas phase.Faraday Discuss. Chem. Soc., 54:257, 1972. ISSN 0301-7249. doi: 10.1039/dc9725400257
-
[52]
J.J. Yeh and I. Lindau. Atomic subshell photoionization cross sections and asymmetry parameters: 1≤Z≤ 103.Atomic Data and Nuclear Data Tables, 32(1):1– 155, January 1985. ISSN 0092640X. doi:10.1016/0092- 640X(85)90016-6
-
[53]
R. P¨ arna, R. Sankari, E. Kukk, E. N˜ ommiste, M. Valden, M. Lastusaari, K. Kooser, K. Kokko, M. Hirsim¨ aki, S. Urpelainen, P. Turunen, A. Kivim¨ aki, V. Pankratov, L. Reisberg, F. Hennies, H. Tarawneh, R. Nyholm, and M. Huttula. FinEstBeaMS – A wide-range Finnish- Estonian Beamline for Materials Science at the 1.5 GeV storage ring at the MAX IV Laborat...
-
[54]
Kuno Kooser, Antti Kivim¨ aki, Paavo Turunen, Rainer P¨ arna, Liis Reisberg, Marco Kirm, Mika Valden, Marko Huttula, and Edwin Kukk. Gas-phase endstation of electron, ion and coincidence spectroscopies for diluted samples at the FinEstBeAMS beamline of the MAX IV 1.5 GeV storage ring.J Synchrotron Rad, 27 (4):1080–1091, July 2020. ISSN 1600-5775. doi: 10....
-
[55]
Kirill Chernenko, Antti Kivim¨ aki, Rainer P¨ arna, Weimin Wang, Rami Sankari, Mats Leandersson, Hamed Tarawneh, Vladimir Pankratov, Mati Kook, Edwin Kukk, Liis Reisberg, Samuli Urpelainen, Tanel K¨ a¨ ambre, Frank Siewert, Grzegorz Gwalt, Andrey Sokolov, Stephanie Lemke, Svyatoslav Alimov, Jeniffa Knedel, Oliver Kutz, Tino Seliger, Mika Valden, Mika Hirs...
-
[56]
Pedro F. Tavares, Simon C. Leemann, Magnus Sj¨ ostr¨ om, and ˚Ake Andersson. The MAX IV storage ring project.J Synchrotron Rad, 21(5):862–877, September 2014. ISSN 1600-5775. doi:10.1107/S1600577514011503
-
[57]
Nils Martensson and Mikael Eriksson. The saga of MAX IV, the first multi-bend achromat synchrotron light source.Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 907:97–104, November 2018. ISSN 01689002. doi: 10.1016/j.nima.2018.03.018
-
[58]
Aymeric Robert, Yngve Cerenius, Pedro Fernandes Tavares, Anna Hultin Stigenberg, Olof Karis, Ann- Christine Lloyd Whelan, Caroline Run´ eus, and Marjolein Thunnissen. MAX IV Laboratory.Eur. Phys. J. Plus, 138(6):495, June 2023. ISSN 2190-5444. doi: 10.1140/epjp/s13360-023-04018-w
-
[59]
Near- threshold photoionization of the Ar 2p subshell.J
L Avaldi, G Dawber, R Camilloni, G C King, M Roper, M R F Siggel, G Stefani, and M Zitnik. Near- threshold photoionization of the Ar 2p subshell.J. Phys. B: At. Mol. Opt. Phys., 27(17):3953–3966, September
-
[60]
QUANTUM ESPRESSO: A modular and open -source software project for quantum simulations of materials
ISSN 0953-4075, 1361-6455. doi:10.1088/0953- 4075/27/17/019
-
[61]
W.L. Jolly, K.D. Bomben, and C.J. Eyermann. Core-electron binding energies for gaseous atoms and molecules.Atomic Data and Nuclear Data Tables, 31 (3):433–493, November 1984. ISSN 0092640X. doi: 10.1016/0092-640X(84)90011-1
-
[62]
L. Karlsson, L. Mattsson, R. Jadrny, R. G. Albridge, S. Pinchas, T. Bergmark, and K. Siegbahn. Isotopic and vibronic coupling effects in the valence electron spectra of H2 16O, H2 18O, and D2 16O.The Journal of Chemical Physics, 62(12):4745–4752, June 1975. ISSN 0021-9606, 1089-7690. doi:10.1063/1.430423
-
[63]
H. Bock. K. Kimura, S. Katsumata, Y. Achiba, T. Yamazaki and S. Iwata: Handbook of He Photoelectron Spectra of Fundamental Organic Molecules. Ionization Energies, Ab Initio Assignments, and Valence Electronic Structure for 200 Molecules. Japan Scientific Societies Press, Tokyo und Halstead Press, New York 1981. 268 Seiten, Preis:$39.95 oder£29.60.Ber Buns...
-
[64]
William L. Jolly and Theodore F. Schaaf. .pi.- Donor relaxation in the oxygen 1s ionization of carbonyl compounds.J. Am. Chem. Soc., 98(11):3178– 3181, May 1976. ISSN 0002-7863, 1520-5126. doi: 10.1021/ja00427a020
-
[66]
Albert A. Bakke, William L. Jolly, and Theodore F. Schaaf. A reversal of the usual binding energy-vs.- charge relationship. The carbon 1s binding energies of bis (cyclopentadienyl) metal complexes in the gas.Journal of Electron Spectroscopy and Related Phenomena, 11 (3):339–342, January 1977. ISSN 03682048. doi: 10.1016/0368-2048(77)80009-1
-
[67]
D.A. Allison, G. Johansson, C.J. Allan, U. Gelius, H. Siegbahn, J. Allison, and K. Siegbahn. Molecular spectroscopy by means of ESCA.Journal of Electron Spectroscopy and Related Phenomena, 1(3):269–283, January 1972. ISSN 03682048. doi:10.1016/0368- 2048(72)85016-3
-
[68]
Ronald G. Cavell and David A. Allison. Site of protonation in aromatic and acyclic amines and acyclic amides revealed by N1s core level electron spectroscopy. J. Am. Chem. Soc., 99(12):4203–4204, June 1977. ISSN 0002-7863, 1520-5126. doi:10.1021/ja00454a072
-
[69]
R. S. Brown and A. Tse. Determination of circumstances under which the correlation of core binding energy and gas-phase basicity or proton affinity breaks down.J. Am. 10 Chem. Soc., 102(16):5222–5226, July 1980. ISSN 0002- 7863, 1520-5126. doi:10.1021/ja00536a017
-
[70]
Giovanna Fronzoni, Oscar Baseggio, Mauro Stener, Weijie Hua, Guangjun Tian, Yi Luo, Barbara Apicella, Michela Alf´ e, Monica De Simone, Antti Kivim¨ aki, and Marcello Coreno. Vibrationally resolved high- resolution NEXAFS and XPS spectra of phenanthrene and coronene.The Journal of Chemical Physics, 141 (4):044313, July 2014. ISSN 0021-9606, 1089-7690. doi...
-
[71]
Si Fen Xiang, Hsiang Wen Chen, Charles J. Eyermann, William L. Jolly, Steven P. Smit, Klaus H. Theopold, Robert G. Bergman, Wolfgang A. Herrmann, and Rowland Pettit. An x-ray photoelectron spectroscopic study of transition metal .mu.-methylene complexes and related compounds.Organometallics, 1(9):1200–1203, September 1982. ISSN 0276-7333, 1520-6041. doi: ...
-
[72]
Steven C. Avanzino, Albert A. Bakke, Hsiang-Wen Chen, Craig J. Donahue, William L. Jolly, Ting Ho Lee, and Antonio J. Ricco. Study of charge transfer in back- bonding to carbonyl and nitrosyl groups.Inorg. Chem., 19(7):1931–1936, July 1980. ISSN 0020-1669, 1520-510X. doi:10.1021/ic50209a022
-
[73]
H.-W. Chen, W.L. Jolly, S.-F. Xiang, I.S. Butler, and J. Sedman. An X-ray photoelectron spectroscopic study of transition-metal thiocarbonyl and thionitrosyl complexes.Journal of Electron Spectroscopy and Related Phenomena, 24(1):121–124, 1981. ISSN 03682048. doi: 10.1016/0368-2048(81)80051-5
-
[74]
David C. Calabro, John L. Hubbard, Charles H. Blevins, Andrew C. Campbell, and Dennis L. Lichtenberger. Effects of methyl group substitution on metal-coordinated cyclopentadienyl rings. Core and valence ionizations of methylated tricarbonyl(.eta.5- cyclopentadienyl)metal complexes.J. Am. Chem. Soc., 103(23):6839–6846, November 1981. ISSN 0002-7863, 1520-5...
-
[75]
Richard R. Rietz, Theodore F. Schaaf, and William L. Jolly. An x-ray photoelectron spectroscopic study of volatile vanadium compounds.Inorg. Chem., 14(11): 2818–2821, November 1975. ISSN 0020-1669, 1520-510X. doi:10.1021/ic50153a044
-
[76]
Marta Berholts, Tanel K¨ a¨ ambre, Arvo T˜ onisoo, Rainer P¨ arna, Vambola Kisand, and Juhan Matthias Kahk. Supplementary data for ”Does the total energy difference method for modelling core level photoemission fail for bigger molecules?”, March 2026. URLhttps://zenodo. org/doi/10.5281/zenodo.19256190
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.