Recognition: 2 theorem links
· Lean TheoremBond-Strength-Based Understanding of Oxygen Vacancy Migration Barriers in Rutile Oxides
Pith reviewed 2026-05-10 17:54 UTC · model grok-4.3
The pith
The average of covalent and ionic bond strengths predicts oxygen vacancy migration barriers in rutile oxides.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
In rutile-type 3d transition-metal dioxides the migration barrier E_B of oxygen vacancies correlates strongly with the covalent contribution S_c obtained from the sum of integrated crystal orbital Hamilton populations and with the ionic contribution S_i obtained from the Madelung energy. Their average bar S gives a reasonable estimate of the DFT barrier E_B^DFT throughout the series. Two parameters extracted by fitting to a large dataset of these dioxides, following the bond-valence model, then permit efficient estimation of E_B.
What carries the argument
The average bar S of covalent bond strength S_c from integrated crystal orbital Hamilton population and ionic bond strength S_i from Madelung energy, which acts as a single metric to estimate the oxygen-vacancy migration barrier.
If this is right
- The migration barrier height can be estimated directly from the average bond-strength metric without new full DFT runs for each oxide.
- Both the covalent and the ionic parts of bonding contribute to setting the barrier, so changes in either affect vacancy motion.
- Once the two parameters are fixed from the 3d dioxide series, barriers for other compositions in the same family become inexpensive to obtain.
- The approach supplies a bond-valence route to screen rutile oxides for desired vacancy mobility.
Where Pith is reading between the lines
- If the same average bond-strength relation holds for rutile oxides outside the 3d row, the method could extend to faster screening of vacancy-mediated diffusion in a wider range of materials.
- The finding implies that deliberately weakening overall bonding, for instance by selective doping, should lower migration barriers and increase oxygen mobility.
- A direct test would be to measure actual ionic conductivity or tracer diffusion rates in one of the studied oxides and check whether the ordering of barriers matches the ordering of the computed bar S values.
Load-bearing premise
The two parameters fitted only to 3d transition-metal dioxides reflect general bond-strength control of the barrier rather than patterns limited to that specific set of materials.
What would settle it
Computing the DFT migration barrier for one additional rutile oxide outside the fitted dataset and finding that the value deviates substantially from the prediction based on bar S or the two fitted parameters would show the estimation method does not hold.
Figures





read the original abstract
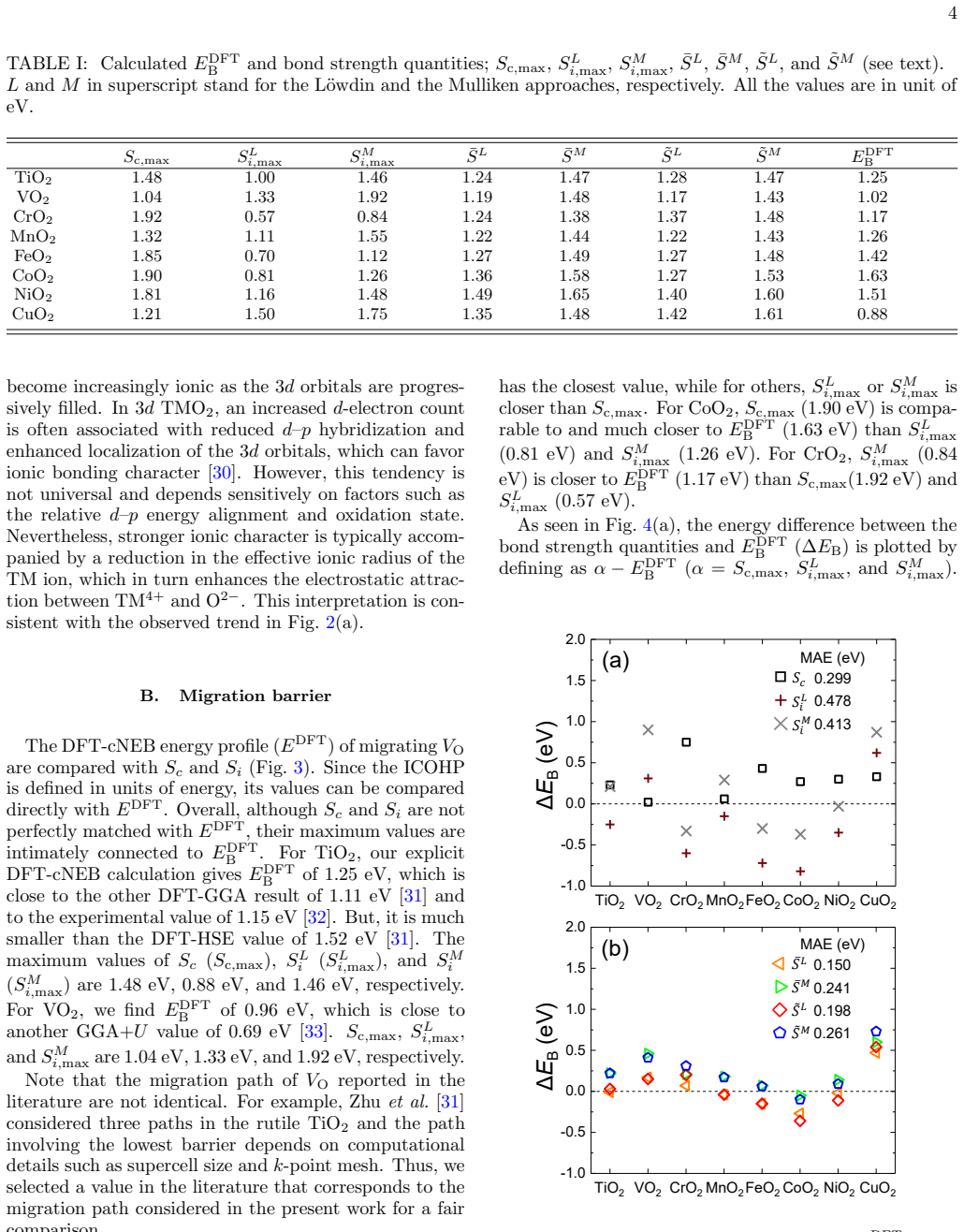
We carry out bond-strength based analysis for the migration barrier ($E_{\rm B}$) of oxygen vacancies in rutile-type 3$d$ transition-metal dioxides by combining density-functional theory (DFT) and the bond-valence model. The covalent and ionic contributions to chemical bonding are explicitly decomposed and quantified by the sum of the integrated crystal orbital Hamilton population ($S_c$) and the Madelung energy ($S_i$), respectively. Both $S_c$ and $S_i$ exhibit strong correlations with the $E_{\rm B}$ from DFT ($E_{\rm B}^{\rm DFT}$), and their average $\bar{S}$ provides a reasonable estimate of $E_{\rm B}^{\rm DFT}$ across the oxide series. Inspired by the bond-valence model, two parameters are extracted by fitting to a large dataset of 3$d$ transition-metal dioxides. Our results show that using these parameters, $E_{\rm B}$ of oxygen vacancies can be efficiently estimated.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that oxygen vacancy migration barriers (E_B) in rutile-type 3d transition-metal dioxides can be understood and efficiently estimated via a bond-strength decomposition. Using DFT, the authors quantify covalent bonding via the sum of integrated crystal orbital Hamilton populations (S_c) and ionic bonding via Madelung energies (S_i). Both quantities correlate strongly with E_B^DFT; their average (with two parameters fitted to the dataset) is presented as a reasonable proxy for E_B across the series, enabling fast estimation without full barrier calculations.
Significance. If the reported correlations prove robust and the fitted estimation generalizes, the work would offer a useful, low-cost alternative to direct DFT barrier computations for defect migration in this class of oxides, while providing physical insight through the explicit covalent/ionic decomposition. The bond-valence-inspired approach and use of a sizable dataset of 3d dioxides are positive elements that could aid materials design for controlled ionic conductivity.
major comments (2)
- [Abstract] Abstract: the central claim that S_c and S_i 'exhibit strong correlations' with E_B^DFT and that their average 'provides a reasonable estimate' is unsupported by any numerical values (correlation coefficients, R², MAE, dataset size, or error bars). This is load-bearing because the abstract supplies no quantitative basis for assessing how well the proxy performs or whether it justifies the 'efficient estimation' conclusion.
- [Abstract] Abstract: the two parameters are 'extracted by fitting to a large dataset of 3d transition-metal dioxides' and then used to estimate E_B on the same series. This creates a circularity risk for the claim of transferable physics; the manuscript must demonstrate out-of-sample validation, sensitivity to DFT functional, or an independent (non-fitted) derivation of the parameters to establish that the estimate reflects causal bond-strength control rather than dataset-specific interpolation.
minor comments (1)
- [Abstract] Abstract: the symbol S-bar is introduced without an explicit definition or formula in the abstract itself, which reduces immediate clarity for readers.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and positive assessment of the potential utility of our bond-strength approach. We address both major comments on the abstract below and have made revisions to strengthen the quantitative presentation and address transferability concerns.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that S_c and S_i 'exhibit strong correlations' with E_B^DFT and that their average 'provides a reasonable estimate' is unsupported by any numerical values (correlation coefficients, R², MAE, dataset size, or error bars). This is load-bearing because the abstract supplies no quantitative basis for assessing how well the proxy performs or whether it justifies the 'efficient estimation' conclusion.
Authors: We agree that the abstract should be self-contained and provide quantitative support for the central claims. The correlations and performance metrics are already quantified in the main text (Section III, Figure 2, and Table I), but we acknowledge that the abstract as originally written did not include them. In the revised manuscript we have updated the abstract to report the Pearson correlation coefficients for S_c and S_i versus E_B^DFT, the associated R² values, the size of the 3d dioxide dataset, and the mean absolute error of the two-parameter estimate, together with a brief statement of the error bars obtained from the DFT calculations. revision: yes
-
Referee: [Abstract] Abstract: the two parameters are 'extracted by fitting to a large dataset of 3d transition-metal dioxides' and then used to estimate E_B on the same series. This creates a circularity risk for the claim of transferable physics; the manuscript must demonstrate out-of-sample validation, sensitivity to DFT functional, or an independent (non-fitted) derivation of the parameters to establish that the estimate reflects causal bond-strength control rather than dataset-specific interpolation.
Authors: We appreciate the referee’s concern about possible circularity. The two parameters have a direct physical interpretation within the bond-valence framework (one weighting the covalent contribution S_c and the other the ionic contribution S_i), so the functional form itself is not fitted ad hoc. Nevertheless, to demonstrate that the estimate is not merely an interpolation of the training set, we have added a leave-one-out cross-validation analysis in the revised manuscript. When each compound is successively excluded from the fit, the mean absolute error remains comparable to the full-dataset value, indicating robustness. We have also included a brief comparison of results obtained with PBE versus HSE06 for a representative subset of the oxides, confirming that the trends and parameter values are not strongly functional-dependent. These additions are placed in the main text and supplementary information. revision: yes
Circularity Check
E_B estimation uses parameters fitted directly to the same DFT dataset of 3d transition-metal dioxides
specific steps
-
fitted input called prediction
[Abstract]
"Inspired by the bond-valence model, two parameters are extracted by fitting to a large dataset of 3$d$ transition-metal dioxides. Our results show that using these parameters, $E_{rm B}$ of oxygen vacancies can be efficiently estimated."
The two parameters are obtained by fitting to the DFT-computed E_B values for the same series of 3d transition-metal dioxides. The subsequent 'efficient estimation' of E_B via the average of S_c and S_i (or the fitted combination) therefore reproduces patterns from the training data by construction, rather than providing an a priori prediction transferable beyond the fitted set.
full rationale
The paper computes S_c (integrated COHP) and S_i (Madelung energy) from DFT, shows their correlation with E_B^DFT, and states that their average provides a reasonable estimate. However, the practical claim of efficient estimation rests on two parameters extracted by fitting to the identical dataset of 3d dioxides. This reduces the reported 'estimate' to a fitted interpolation on the input E_B^DFT values rather than an independent derivation from bond-valence theory. No out-of-sample validation or parameter-free derivation is indicated in the abstract or description. The core correlation analysis is independent, but the load-bearing estimation step is not.
Axiom & Free-Parameter Ledger
free parameters (1)
- two fitted parameters
axioms (1)
- domain assumption Bond-valence model quantities (Madelung energy) correctly capture ionic bonding contribution in these rutile oxides
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
two parameters are extracted by fitting to a large dataset of 3d transition-metal dioxides... E_B of oxygen vacancies can be efficiently estimated
-
IndisputableMonolith/Foundation/AlphaCoordinateFixation.leanJ_uniquely_calibrated_via_higher_derivative unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Inspired by the bond-valence model... r0^ICOHP and b^ICOHP are obtained using the 795 data set
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
stretching
B. Yildiz, “stretching” the energy landscape of ox- ides—effects on electrocatalysis and diffusion, MRS Bull. 39, 147 (2014)
2014
-
[2]
Waser and M
R. Waser and M. Aono, Nanoionics-based resistive switching memories, Nat. Mater.6, 833 (2007)
2007
-
[3]
Freysoldt, B
C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti, and C. G. Van de Walle, First- principles calculations for point defects in solids, Rev. Mod. Phys.86, 253 (2014)
2014
-
[4]
C. G. Van de Walle and J. Neugebauer, First-principles calculations for defects and impurities: Applications to iii-nitrides, J. Appl. Phys.95, 3851 (2004)
2004
-
[5]
F. Oba, M. Choi, A. Togo, and I. Tanaka, Point defects in zno: an approach from first principles, Sci. Technol. Adv. Mater.12, 034302 (2011)
2011
-
[6]
Limpijumnong and C
S. Limpijumnong and C. G. Van de Walle, Diffusivity of native defects in gan, Phys. Rev. B69, 035207 (2004)
2004
-
[7]
Mayeshiba and D
T. Mayeshiba and D. Morgan, Strain effects on oxygen migration in perovskites, Phys. Chem. Chem. Phys.17, 2715 (2015)
2015
-
[8]
T. T. Mayeshiba and D. D. Morgan, Factors controlling oxygen migration barriers in perovskites, Solid State Ion- ics296, 71 (2016)
2016
-
[9]
Alaydrus, I
M. Alaydrus, I. Hamada, and Y. Morikawa, Mechanistic insight into oxygen vacancy migration in srfeo 3-δfrom dft+ u simulations, Phys. Chem. Chem. Phys.23, 18628 (2021)
2021
-
[10]
S. Zhao, L. Gao, C. Lan, S. S. Pandey, S. Hayase, and T. Ma, Oxygen vacancy formation and migration in dou- ble perovskite sr2crmoo6: a first-principles study, RSC Adv.6, 43034 (2016)
2016
-
[11]
Henkelman, B
G. Henkelman, B. P. Uberuaga, and H. J´ onsson, A climb- ing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys.113, 9901 (2000)
2000
-
[12]
R. Devi, B. Singh, P. Canepa, and G. Sai Gautam, Ef- fect of exchange-correlation functionals on the estimation of migration barriers in battery materials, npj Comput. Mater.8, 160 (2022)
2022
-
[13]
J. A. Santana, J. T. Krogel, P. R. Kent, and F. A. Re- boredo, Diffusion quantum monte carlo calculations of sr- feo3 and lafeo3, J. Chem. Phys.147, 10.1063/1.4990667 (2017)
-
[14]
X. Yang, A. J. Fern´ andez-Carri´ on, and X. Kuang, Oxide ion-conducting materials containing tetrahedral moieties: Structures and conduction mechanisms, Chem. Rev.123, 9356 (2023)
2023
-
[15]
Dronskowski and P
R. Dronskowski and P. E. Bloechl, Crystal orbital hamil- ton populations (cohp): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem.97, 8617 (1993)
1993
-
[16]
V. Fung, Z. Wu, and D.-e. Jiang, New bonding model of radical adsorbate on lattice oxygen of perovskites, J. Phys. Chem. Lett.9, 6321 (2018)
2018
-
[17]
I. Kim, H. Lee, and M. Choi, First-principles study of oxygen vacancy formation in strained oxides, J. Appl. Phys.131, 10.1063/5.0084795 (2022)
-
[19]
I. D. Brown, Recent developments in the methods and applications of the bond valence model, Chem. Rev.109, 6858 (2009)
2009
-
[20]
Adams and R
S. Adams and R. P. Rao, Transport pathways for mo- bile ions in disordered solids from the analysis of energy- scaled bond-valence mismatch landscapes, Phys. Chem. Chem. Phys.11, 3210 (2009)
2009
-
[21]
Adams and R
S. Adams and R. P. Rao, High power lithium ion battery materials by computational design, Phys. Status Solidi A 208, 1746 (2011)
2011
-
[22]
P. E. Bl¨ ochl, Projector augmented-wave method, Phys. Rev. B50, 17953 (1994)
1994
-
[23]
Kresse and J
G. Kresse and J. Hafner, Ab initio molecular dynamics for open-shell transition metals, Phys. Rev. B48, 13115 (1993)
1993
-
[24]
V. L. Deringer, A. L. Tchougreff, and R. Dronskowski, Crystal orbital hamilton population (cohp) analysis as projected from plane-wave basis sets, J. Phys. Chem. A 115, 5461 (2011)
2011
-
[25]
P. C. Muller, C. Ertural, J. Hempelmann, and R. Dron- skowski, Crystal orbital bond index: covalent bond or- ders in solids, J. Phys. Chem. C125, 7959 (2021)
2021
-
[26]
S. P. Ong, W. D. Richards, A. Jain, G. Hautier, 9 M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson, and G. Ceder, Python materials genomics (py- matgen): A robust, open-source python library for ma- terials analysis, Comput. Mater. Sci.68, 314 (2013)
2013
-
[27]
R. G. Pearson, Absolute electronegativity and hardness: application to inorganic chemistry, Inorganic chemistry 27, 734 (1988)
1988
-
[28]
R. D. Shannon, Revised effective ionic radii and sys- tematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr. Sect. A32, 751 (1976)
1976
-
[29]
Adams, Relationship between bond valence and bond softness of alkali halides and chalcogenides, Acta Crys- tallographica Section B57, 278 (2001)
S. Adams, Relationship between bond valence and bond softness of alkali halides and chalcogenides, Acta Crys- tallographica Section B57, 278 (2001)
2001
-
[30]
C.-Y. Liu, L. Celiberti, R. Decker, K. Ruotsalainen, K. Siewierska, M. Kusch, R.-P. Wang, D. J. Kim, I. I. Olaniyan, D. Di Castro,et al., Orbital-overlap-driven hybridization in 3d-transition metal perovskite oxides lamo3 (m= ti-ni) and la2cuo4, Commun. Phys.7, 156 (2024)
2024
-
[31]
Zhu, Q.-M
L. Zhu, Q.-M. Hu, and R. Yang, The effect of electron lo- calization on the electronic structure and migration bar- rier of oxygen vacancies in rutile, Journal of Physics: Condensed Matter26, 055602 (2014)
2014
-
[32]
Zhang, Q
Z. Zhang, Q. Ge, S.-C. Li, B. D. Kay, J. M. White, and Z. Dohn´ alek, Imaging intrinsic diffusion of bridge- bonded oxygen vacancies on tio 2(110), Phys. Rev. Lett. 99, 126105 (2007)
2007
-
[33]
Sim, K.-Y
H. Sim, K.-Y. Doh, Y. Park, K. Song, G.-Y. Kim, J. Son, D. Lee, and S.-Y. Choi, Crystallographic pathways to tai- loring metal-insulator transition through oxygen trans- port in vo2, Small20, 2402260 (2024)
2024
-
[34]
O. C. Gagn´ e and F. C. Hawthorne, Comprehensive derivation of bond-valence parameters for ion pairs in- volving oxygen, Acta Crystallogr. Sect. B71, 562 (2015)
2015
-
[35]
I. D. Brown and D. Altermatt, Bond-valence parameters obtained from a systematic analysis of the inorganic crys- tal structure database, Acta Crystallogr. Sect. B41, 244 (1985)
1985
-
[36]
J. C. Slater, Atomic shielding constants, Physical Review 36, 57 (1930)
1930
-
[37]
J. K. Burdett and F. C. Hawthorne, An orbital approach to the theory of bond valence, American Mineralogist78, 884 (1993)
1993
-
[38]
de Jong,et al., Charting the complete elastic properties of inorganic crystalline compounds
M. de Jong, W. Chen, T. Angsten, A. Jain, R. Notes- tine, A. Gamst, M. Sluiter, C. Krishna Ande, S. van der Zwaag, J. J. Plata, C. Toher, S. Curtarolo, G. Ceder, K. A. Persson, and M. Asta, Charting the complete elas- tic properties of inorganic crystalline compounds, Sci. Data2, 10.1038/sdata.2015.9 (2015)
-
[39]
Adams, Relationship between bond valence and bond softness of alkali halides and chalcogenides, Structural Science57, 278 (2001)
S. Adams, Relationship between bond valence and bond softness of alkali halides and chalcogenides, Structural Science57, 278 (2001)
2001
-
[40]
Geerlings, F
P. Geerlings, F. De Proft, and W. Langenaeker, Con- ceptual density functional theory, Chem. Rev.103, 1793 (2003)
2003
-
[41]
Soulard, X
C. Soulard, X. Rocquefelte, S. Jobic, D. Dai, H.-J. Koo, and M.-H. Whangbo, Metal–ligand bonding and rutile- versus cdi2-type structural preference in platinum diox- ide and titanium dioxide, Journal of Solid State Chem- istry175, 353 (2003)
2003
-
[42]
Toropova, G
A. Toropova, G. Kotliar, S. Y. Savrasov, and V. S. Oudovenko, Electronic structure and magnetic anisotropy of cro 2, Phys. Rev. B71, 172403 (2005)
2005
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.