Recognition: unknown
A Massively Scalable Ligand-Protein Dissociation Dynamic Database Derived from Atomistic Molecular Modelling
Pith reviewed 2026-05-10 17:36 UTC · model grok-4.3
The pith
A database supplies atomistic dissociation trajectories for 19,037 ligand-protein complexes and assigns their rate constants.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
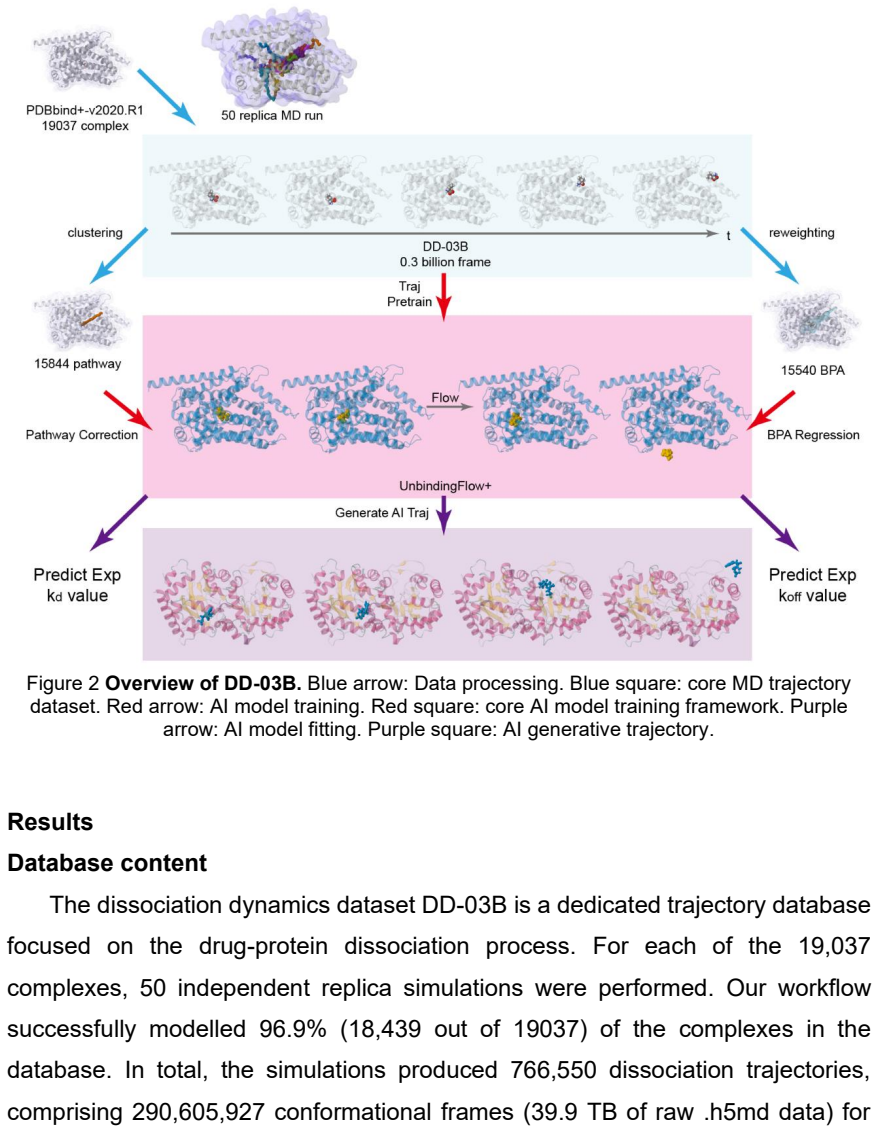
Utilising and extending a validated computational pipeline, dissociation trajectories were generated for 19,037 ligand-protein complexes sourced from PDBbind+v2020R1, resulting in a repository of approximately 0.3 billion simulation frames. For these systems—which possess experimental binding affinities but typically lack measured koff rates—dissociation rate constants were computed and assigned through trajectory reweighting. Analysis reveals that protein-ligand complexes fall into three mechanistic types (pathway-dominant, open-pocket, and entropy-pocket systems), each requiring distinct strategies for accurate kinetic characterisation. Together with prior data, the collection forms the DD
What carries the argument
The extended atomistic molecular modelling pipeline that generates dissociation trajectories and applies trajectory reweighting to assign dissociation rate constants.
If this is right
- The database supplies raw dynamic data that can train and benchmark generative AI models for predicting drug-protein dissociation kinetics.
- Complexes are grouped into pathway-dominant, open-pocket, and entropy-pocket types, each needing tailored methods for kinetic characterisation.
- The collection becomes the core of an expandable Dissociation Dynamic Database that will grow with additional trajectories.
- Public release of the 40 TB data set enables community-wide use for computational drug discovery focused on kinetics.
Where Pith is reading between the lines
- Large-scale access to simulated unbinding paths could shift drug design emphasis from static affinity to controlled dissociation speed.
- Linking the mechanistic categories to clinical success rates might show which type of complex is most common among approved drugs.
- Adding simulations that vary temperature or solvent conditions would test whether the three-type classification remains stable.
Load-bearing premise
The computational pipeline remains accurate for dissociation trajectories across the full set of complexes and trajectory reweighting can reliably assign koff values where no experimental measurements exist.
What would settle it
New experimental dissociation rate measurements for a representative subset of the complexes, followed by direct numerical comparison to the rates assigned by trajectory reweighting.
Figures
read the original abstract
Understanding the kinetics of drug-protein interactions is paramount for drug design, yet the field lacks large-scale, dynamic data to move beyond static structural analysis. Here, we present DD-03B, a massively scalable database providing dynamic, all-atom dissociation trajectories for a broad set of ligand-protein complexes. Utilising and extending a validated computational pipeline, we generated dissociation trajectories for 19,037 ligand-protein complexes sourced from PDBbind+v2020R1, resulting in a repository of approximately 0.3 billion simulation frames totalling 40 TB in size. For these systems-which possess experimental binding affinities (kd) but typically lack measured koff rates-we computed and assigned dissociation rate constants through trajectory reweighting. Our analysis reveals that protein-ligand complexes can be categorised into three mechanistic types (pathway-dominant, open-pocket, and entropy-pocket systems), each requiring distinct strategies for accurate kinetic characterisation. Together with our previously released DD-13M, DD-03B forms the core of the expandable Dissociation Dynamic Database (DDD) project, which will be continuously augmented with new trajectories. This large-scale, publicly available resource establishes a critical foundation for training and benchmarking next-generation generative AI models to predict and optimise drug-protein dissociation kinetics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents DD-03B, a database of all-atom dissociation trajectories for 19,037 ligand-protein complexes sourced from PDBbind v2020R1. The authors report generating approximately 0.3 billion simulation frames (40 TB) using an extended validated computational pipeline, then assigning dissociation rate constants (koff) to these systems via trajectory reweighting despite most complexes lacking experimental koff data (only kd available). They further categorize the complexes into three mechanistic types—pathway-dominant, open-pocket, and entropy-pocket systems—and position the resource as the core of an expandable Dissociation Dynamic Database (DDD) project intended for training generative AI models on dissociation kinetics.
Significance. A rigorously validated large-scale dynamic database of dissociation trajectories would be a significant contribution to computational drug design, as it could supply the missing kinetic dimension beyond static structures and enable data-driven models for koff prediction. The proposed mechanistic categorization, if substantiated with clear criteria and reproducible analysis, could also provide conceptual value for understanding dissociation pathways. However, the absence of quantitative validation metrics means the claimed utility for AI training and benchmarking remains conditional on future demonstration that the reweighting yields transferable, non-circular predictions.
major comments (3)
- [Abstract] Abstract: The central claim that koff values were 'computed and assigned ... through trajectory reweighting' for the 19,037 complexes is unsupported by any quantitative validation metrics, error estimates, or direct comparisons against experimental koff values (even on the small subset where such data exist). This directly affects whether the assigned rates and the three mechanistic categories can be treated as reliable inputs for downstream AI training.
- [Abstract] Abstract: The reweighting procedure used to obtain koff from trajectories is described only at the level of the abstract; no details are supplied on the functional form, any free parameters, the reference data employed, or how transferability to complexes lacking experimental koff was assessed. Without these, it is impossible to evaluate whether the procedure is independent of the kd data used for validation or whether it reduces to a fitted quantity.
- [Abstract] Abstract: The statement that protein-ligand complexes 'can be categorised into three mechanistic types' is presented without any description of the classification criteria, the analysis performed on the trajectories, or quantitative support (e.g., clustering statistics or separation metrics). This categorization is load-bearing for the claim that distinct strategies are required for accurate kinetic characterisation.
minor comments (2)
- The manuscript should include a dedicated methods or results subsection that reports the size of the experimental koff validation set, the numerical agreement (e.g., correlation coefficient, mean absolute error) between reweighted and measured koff, and any cross-validation strategy used to test transferability.
- Clarify the relationship between the previously released DD-13M and the new DD-03B datasets, including any overlap in complexes and how the combined DDD resource will be versioned and accessed.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. We address each major comment below and will revise the manuscript to incorporate additional details and quantitative support as outlined.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that koff values were 'computed and assigned ... through trajectory reweighting' for the 19,037 complexes is unsupported by any quantitative validation metrics, error estimates, or direct comparisons against experimental koff values (even on the small subset where such data exist). This directly affects whether the assigned rates and the three mechanistic categories can be treated as reliable inputs for downstream AI training.
Authors: We agree that the abstract lacks explicit quantitative validation metrics and error estimates. The full manuscript describes the reweighting approach applied to systems with kd data and notes the scarcity of experimental koff values. In revision, we will add a dedicated validation subsection reporting comparisons to experimental koff on the available subset, along with error estimates and metrics assessing reliability for AI training. The abstract will be updated to reference these additions. revision: yes
-
Referee: [Abstract] Abstract: The reweighting procedure used to obtain koff from trajectories is described only at the level of the abstract; no details are supplied on the functional form, any free parameters, the reference data employed, or how transferability to complexes lacking experimental koff was assessed. Without these, it is impossible to evaluate whether the procedure is independent of the kd data used for validation or whether it reduces to a fitted quantity.
Authors: We acknowledge the need for greater methodological transparency. While the methods section outlines the overall pipeline, we will expand it in the revision to specify the exact functional form of the reweighting, all free parameters, the reference data sources, and the assessment of transferability to systems without experimental koff. The abstract will be revised to direct readers to these details, clarifying independence from kd validation data. revision: yes
-
Referee: [Abstract] Abstract: The statement that protein-ligand complexes 'can be categorised into three mechanistic types' is presented without any description of the classification criteria, the analysis performed on the trajectories, or quantitative support (e.g., clustering statistics or separation metrics). This categorization is load-bearing for the claim that distinct strategies are required for accurate kinetic characterisation.
Authors: We will revise the manuscript to include explicit classification criteria, a description of the trajectory analysis used, and quantitative support such as clustering statistics and separation metrics for the three mechanistic types. These additions will appear in the methods and results sections, with the abstract updated to summarize the supporting analysis. revision: yes
Circularity Check
No significant circularity; derivation is self-contained
full rationale
The paper generates dissociation trajectories for 19,037 complexes using an extended validated pipeline and assigns koff via trajectory reweighting on systems that have kd but lack measured koff. The reweighting is presented as a direct computational procedure applied to the new trajectories rather than a fit to the target koff or kd values. The three mechanistic categories are obtained by post-hoc analysis of the resulting trajectories. Self-reference to the prior DD-13M release and the validated pipeline supplies context and prior validation but does not substitute for the current generation or reweighting steps; those steps remain independent computations whose correctness can be checked against external benchmarks or additional experiments. No equation or claim reduces by construction to its own inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The computational pipeline used to generate dissociation trajectories is validated and can be extended without loss of accuracy.
invented entities (1)
-
Three mechanistic types (pathway-dominant, open-pocket, entropy-pocket systems)
no independent evidence
Reference graph
Works this paper leans on
-
[1]
(3) Yim, J.; Stärk, H.; Corso, G.; Jing, B.; Barzilay, R.; Jaakkola, T
DOI: 10.1021/acs.accounts.6b00491. (3) Yim, J.; Stärk, H.; Corso, G.; Jing, B.; Barzilay, R.; Jaakkola, T. S. Diffusion models in protein structure and docking. WIREs Computational Molecular Science 2024, 14 (2), e1711. DOI: https://doi.org/10.1002/wcms.1711. (4) Cao, D.; Chen, M.; Zhang, R.; Wang, Z.; Huang, M.; Yu, J.; Jiang, X.; Fan, Z.; Zhang, W.; Zho...
-
[2]
doi: 10.1038/s43588-024-00627-2
DOI: 10.1038/s43588-024-00627-2. (7) Vander Meersche, Y.; Cretin, G.; Gheeraert, A.; Gelly, J. C.; Galochkina, T. ATLAS: protein flexibility description from atomistic molecular dynamics simulations. Nucleic Acids Res 2024, 52 (D1), D384–d392. DOI: 10.1093/nar/gkad1084 From NLM. (8) Mokhtari, O.; Bignon, E.; Khakzad, H.; Karami, Y. DynaRepo: The repositor...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.