Recognition: unknown
Transferable FB-GNN-MBE Framework for Potential Energy Surfaces: Data-Adaptive Transfer Learning in Deep Learned Many-Body Expansion Theory
Pith reviewed 2026-05-10 16:34 UTC · model grok-4.3
The pith
FB-GNN-MBE combines fragment-based graph neural networks with many-body expansion to predict potential energy surfaces at chemical accuracy while transferring across cluster sizes with limited new data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
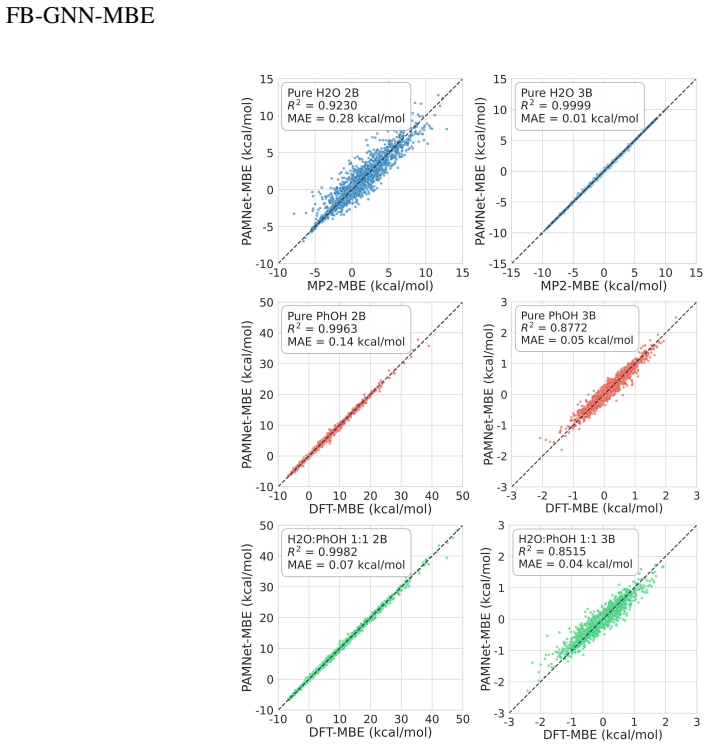
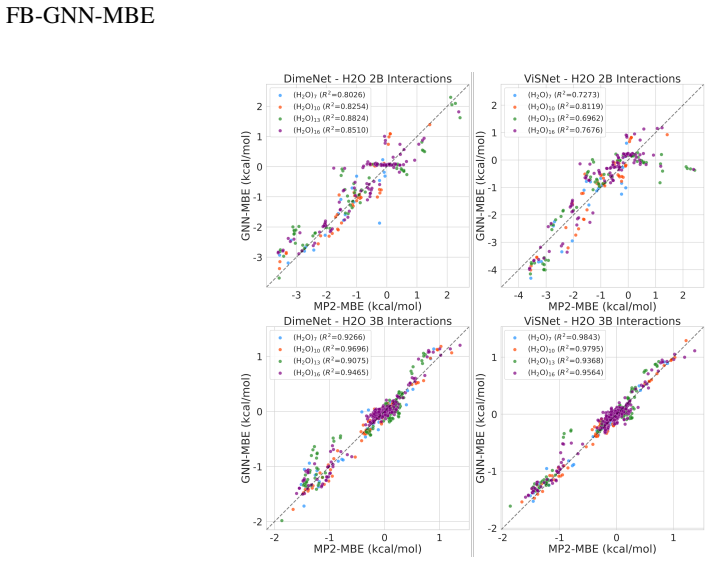
The authors state that FB-GNN-MBE reproduces first-principles potential energy surfaces for hierarchically structured systems with manageable accuracy, complexity, and interpretability. Specifically, the framework achieves chemical accuracy in two-body and three-body energies across water, phenol, and mixture benchmarks as well as the one-dimensional dissociation curves of water and phenol dimers. The teacher-student learning protocol, in which a heavy-weight FB-GNN trained on a mixed-density water cluster ensemble distills knowledge to a light-weight GNN later fine-tuned on a uniform-density (H2O)21 ensemble, produces efficient and accurate two-body and three-body predictions for variously
What carries the argument
Fragment-based graph neural network (FB-GNN) integrated into many-body expansion (MBE) theory, with a teacher-student distillation protocol that transfers learned many-fragment interactions from a heavy model on mixed-density data to a light model fine-tuned on uniform-density clusters.
If this is right
- FB-GNN-MBE predicts two-body and three-body energies to chemical accuracy for water, phenol, and mixture benchmarks.
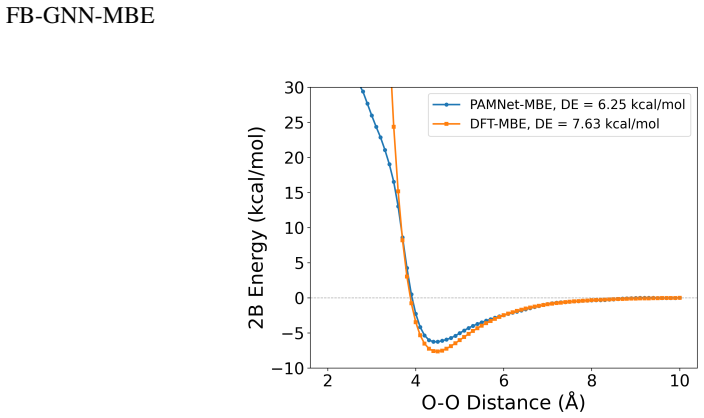
- The framework reproduces one-dimensional dissociation curves for water and phenol dimers.
- The teacher-student protocol yields accurate two- and three-body predictions for water clusters of varying sizes without full retraining.
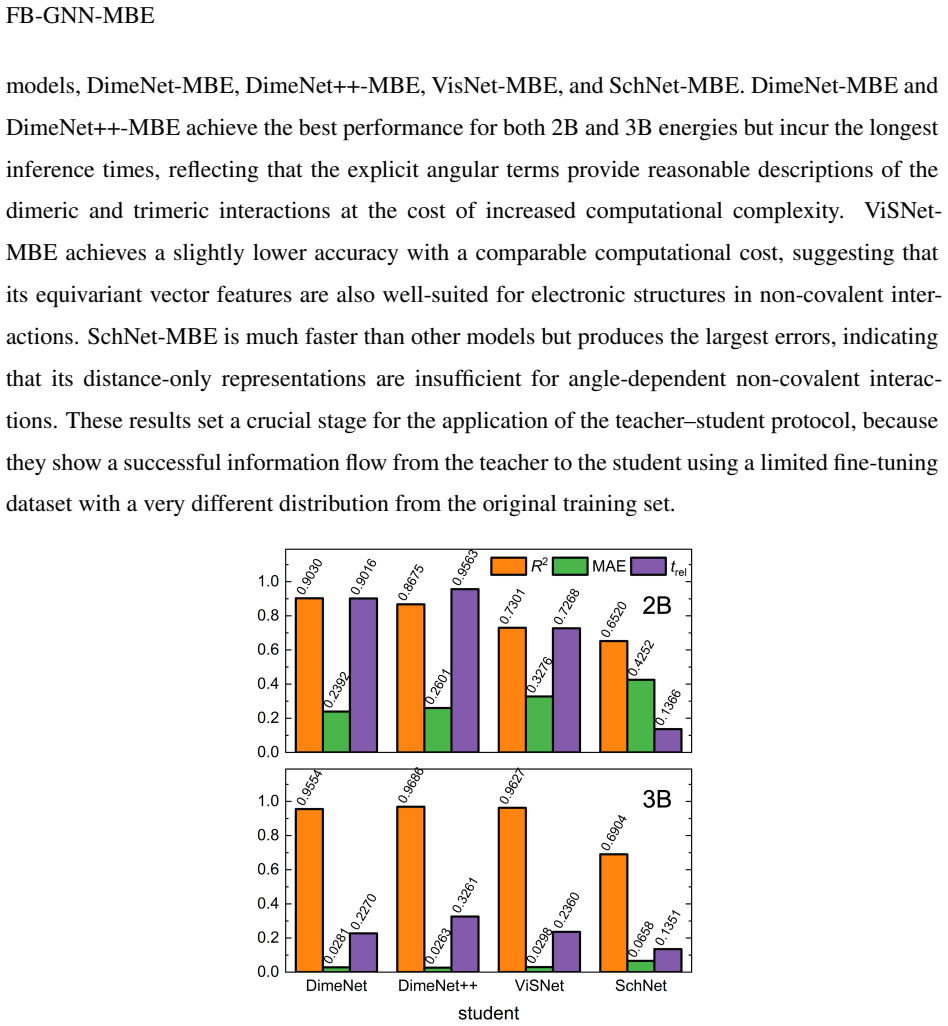
- FB-GNN-MBE outperforms conventional non-fragment GNN models for large-scale molecular simulations.
Where Pith is reading between the lines
- The same distillation step could be applied to adapt the model to other solvents or to mixed molecular environments by changing only the teacher training set.
- If the transfer remains stable, the approach would lower the data-collection cost for modeling extended systems such as solvated biomolecules.
- Explicit addition of four-body terms learned by the same FB-GNN architecture might further reduce errors in dense or long-range regimes.
Load-bearing premise
The fragment-based GNN trained on limited cluster data can generalize many-body interactions to target systems of different sizes and densities without large errors from distribution shift or the need for explicit higher-order terms.
What would settle it
Compute direct quantum-mechanical two- and three-body energies for a water cluster of size outside the training distribution and check whether FB-GNN-MBE deviations exceed chemical accuracy of 1 kcal/mol.
Figures












read the original abstract
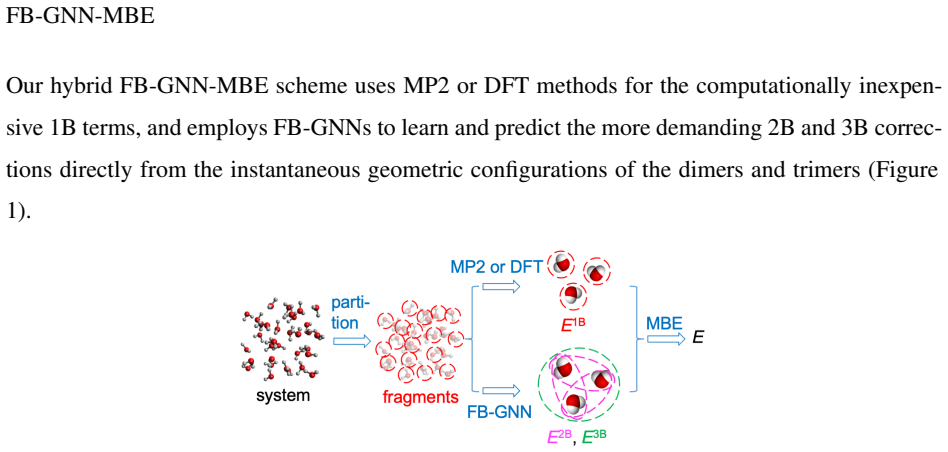
Mechanistic understanding and rational design of complex chemical systems depend on fast and accurate predictions of electronic structures beyond individual building blocks. However, if the system exceeds hundreds of atoms, first-principles quantum mechanical (QM) modeling becomes impractical. In this study, we developed FB-GNN-MBE by integrating a fragment-based graph neural network (FB-GNN) into the many-body expansion (MBE) theory and demonstrated its capacity to reproduce first-principles potential energy surfaces (PES) for hierarchically structured systems with manageable accuracy, complexity, and interpretability. Specifically, we divided the entire system into basic building blocks (fragments), evaluated their one-fragment energies using a QM model, and addressed many-fragment interactions using the structure-property relationships trained by FB-GNNs. Our investigation shows that FB-GNN-MBE achieves chemical accuracy in predicting two-body (2B) and three-body (3B) energies across water, phenol, and mixture benchmarks, as well as the one-dimensional dissociation curves of water and phenol dimers. To transfer the success of FB-GNN-MBE across various systems with minimal computational costs and data demands, we developed and validated a teacher-student learning protocol. A heavy-weight FB-GNN trained on a mixed-density water cluster ensemble (teacher) distills its learned knowledge and passes it to a light-weight GNN (student), which is later fine-tuned on a uniform-density (H2O)21 cluster ensemble. This transfer learning strategy resulted in efficient and accurate prediction of 2B and 3B energies for variously sized water clusters without retraining. Our transferable FB-GNN-MBE framework outperformed conventional non-FB-GNN-based models and showed high practicality for large-scale molecular simulations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces FB-GNN-MBE, which embeds a fragment-based graph neural network into many-body expansion theory to model potential energy surfaces of large systems. It reports that the approach reaches chemical accuracy for 2B and 3B energies on water, phenol, and mixture benchmarks plus dimer dissociation curves, and presents a teacher-student protocol in which a heavy-weight FB-GNN trained on mixed-density water clusters distills knowledge to a light-weight student that is fine-tuned only on uniform-density (H2O)21 clusters, enabling accurate 2B/3B predictions for water clusters of varying sizes without further retraining.
Significance. If the transferability claim is substantiated, the framework would provide a practical route to QM-accurate PES for systems with hundreds of atoms at modest data and compute cost, leveraging MBE interpretability while using GNNs only for the many-body corrections. The teacher-student distillation step is a concrete strength for minimizing data demands when moving between cluster densities and sizes.
major comments (2)
- [Results (transfer learning subsection)] Results section on transfer learning (teacher-student protocol): the assertion that the fine-tuned student reproduces 2B and 3B energies for 'variously sized' water clusters without retraining is load-bearing for the central transferability claim, yet the manuscript supplies no explicit tests of distribution shift in fragment-pair and fragment-triplet geometries (e.g., increased distant pairs or altered coordination numbers) as cluster size grows beyond the (H2O)21 fine-tuning distribution. Without such checks, per-term errors could accumulate in the MBE sum even if small on the training regime.
- [Results (benchmark tables)] Benchmark results (water/phenol/mixture tables): the chemical-accuracy statements are presented without reported error bars, explicit validation splits, data-exclusion criteria, or direct baseline comparisons against non-FB-GNN MBE or standard GNN models, making it impossible to determine whether the reported accuracy is robust or influenced by post-hoc fitting choices.
minor comments (2)
- [Methods and Results] Notation for fragment energies and interaction terms is introduced in the abstract and methods but not consistently cross-referenced in the results figures, reducing readability.
- [Abstract] The abstract states 'outperformed conventional non-FB-GNN-based models' but does not specify which models or metrics were used for the comparison.
Simulated Author's Rebuttal
We thank the referee for their thorough review and insightful comments on our manuscript. We address each of the major comments below and have revised the manuscript accordingly to improve the clarity and robustness of our claims.
read point-by-point responses
-
Referee: [Results (transfer learning subsection)] Results section on transfer learning (teacher-student protocol): the assertion that the fine-tuned student reproduces 2B and 3B energies for 'variously sized' water clusters without retraining is load-bearing for the central transferability claim, yet the manuscript supplies no explicit tests of distribution shift in fragment-pair and fragment-triplet geometries (e.g., increased distant pairs or altered coordination numbers) as cluster size grows beyond the (H2O)21 fine-tuning distribution. Without such checks, per-term errors could accumulate in the MBE sum even if small on the training regime.
Authors: We appreciate the referee's point regarding the need for explicit validation of distribution shifts in the transfer learning protocol. Our current results demonstrate accurate predictions on water clusters of sizes both smaller and larger than the (H2O)21 used for fine-tuning, supporting the transferability without retraining. However, to directly address concerns about potential accumulation of errors due to geometric shifts, we will add a new analysis in the supplementary material. This will include histograms or statistics on key geometric features such as fragment-pair distances and coordination numbers across different cluster sizes, comparing the training distribution to the test distributions. This addition will substantiate that the model generalizes across the observed shifts. revision: yes
-
Referee: [Results (benchmark tables)] Benchmark results (water/phenol/mixture tables): the chemical-accuracy statements are presented without reported error bars, explicit validation splits, data-exclusion criteria, or direct baseline comparisons against non-FB-GNN MBE or standard GNN models, making it impossible to determine whether the reported accuracy is robust or influenced by post-hoc fitting choices.
Authors: We agree that providing more detailed statistical information and baseline comparisons will strengthen the presentation of our benchmark results. In the revised manuscript, we will update the tables to include error bars, which will be obtained from multiple independent training runs with different random seeds. We will also explicitly state the data splitting strategy (e.g., train/validation/test ratios) and any exclusion criteria applied to the datasets. Additionally, we will expand the comparisons by including results from standard GNN models (without fragment-based decomposition) and traditional MBE approaches using fixed functional forms, to clearly highlight the performance gains of the FB-GNN-MBE framework. These revisions will be reflected in the Results section and associated tables. revision: yes
Circularity Check
No significant circularity in FB-GNN-MBE derivation or transfer claims
full rationale
The paper integrates a standard many-body expansion (MBE) with a fragment-based GNN trained on QM fragment energies to approximate 2B/3B interaction terms. The reported chemical accuracy is an empirical validation result obtained by comparing GNN outputs against held-out QM benchmarks on water, phenol, and mixture systems. The teacher-student protocol consists of sequential supervised training stages (mixed-density teacher, then fine-tuning on (H2O)21), followed by evaluation on variously sized clusters; success is measured by external QM agreement rather than by algebraic identity with the training inputs. No equations, definitions, or self-citations in the abstract or described chain reduce any central claim to its own fitted values by construction. The transferability statement is a testable generalization claim, not a self-referential renaming or uniqueness theorem imported from prior author work. This is a conventional data-driven modeling paper whose core results rest on benchmark comparisons, not on internal re-derivation.
Axiom & Free-Parameter Ledger
free parameters (1)
- FB-GNN weights and architecture hyperparameters
axioms (1)
- domain assumption Many-body expansion can be truncated after three-body terms while retaining chemical accuracy for the tested systems
Reference graph
Works this paper leans on
-
[1]
1G. A. Cisneros, K. T. Wikfeldt, L. Ojamäe, J. Lu, Y . Xu, H. Torabifard, A. P . Bartók, G. Csányi, V . Molinero, and F. Paesani, Chemical Reviews 116, 7501 (2016). 2A. S. Christensen, T. Kuba ˇr, Q. Cui, and M. Elstner, Chemical Reviews 116, 5301 (2016). 3L. Rummel and P . R. Schreiner, Angewandte Chemie International Edition 63, e202316364 (2024). 4Y . ...
2016
-
[2]
1025–1035
pp. 1025–1035. 60S. Kearnes, K. McCloskey, M. Berndl, V . Pande, and P . Riley, Journal of Computer-Aided Molecular Design 30, 595 (2016) . 61D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik, and R. P . Adams, in Advances in Neural Information Processing Systems , V ol. 28, edited by C. Cortes, N. Lawrence, D. Lee, M...
2016
-
[3]
2224–2232
pp. 2224–2232. 62C. Chen, W. Y e, Y . Zuo, C. Zheng, and S. P . Ong,Chemistry of Materials 31, 3564 (2019). 63K. Schütt, P .-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko, and K.-R. Müller, in Advances in Neural Information Processing Systems , V ol. 30, edited by I. Guyon, U. V . Luxburg, S. Bengio, H. Wallach, R. Fergus, S. Vishwanathan,...
2019
-
[4]
Margraf, and Stephan Günnemann
pp. 992–1002. 52 FB-GNN-MBE 64K. T. Schütt, H. E. Sauceda, P .-J. Kindermans, A. Tkatchenko, and K.-R. Müller, The Journal of Chemical Physics 148, 241722 (2018). 65O. T. Unke and M. Meuwly, Journal of Chemical Theory and Computation 15, 3678 (2019). 66J. Gasteiger, J. Groß, and S. Günnemann, in International Conference on Learning Represen- tations (Curr...
-
[5]
9323–9332
pp. 9323–9332. 69J. Brandstetter, R. Hesselink, E. van der Pol, E. J. Bekkers, and M. Welling, in International Conference on Learning Representations (Curran Associates, Inc., 2022). 70Y . Wang, T. Wang, S. Li, X. He, M. Li, Z. Wang, N. Zheng, B. Shao, and T.-Y . Liu, Nature Communications 15, 313 (2024) . 71I. Batatia, D. P . Kovacs, G. Simm, C. Ortner,...
2022
-
[6]
pp. 11423–11436. 72S. Zhang, Y . Liu, and L. Xie, NeurIPS’2020 Machine Learning for Structural Biology Work- shop, arXiv Preprint , arXiv:2011.07457 (2020) . 73S. Zhang, Y . Liu, and L. Xie, Scientific Reports 13, 19171 (2023) . 74E. Alsentzer, S. Finlayson, M. Li, and M. Zitnik, in Advances in Neural Information Processing Systems, V ol. 33, edited by H. ...
-
[7]
8017–8029
pp. 8017–8029. 75E. M. Collins and K. Raghavachari, The Journal of Physical Chemistry A 125, 6872 (2021). 76J. Gilmer, S. S. Schoenholz, P . F. Riley, O. Vinyals, and G. E. Dahl, in Proceedings of the 34th International Conference on Machine Learning , Proceedings of Machine Learning Research, V ol. 70, edited by D. Precup and Y . W. Teh (PMLR,
2021
-
[8]
p. 803812. 78X. Wang, D. Bo, C. Shi, S. Fan, Y . Y e, and S. Y . Philip,IEEE Transactions on Big Data 9, 415 (2023). 53 FB-GNN-MBE 79S. Chen, Z. Wang, X. Deng, Y . Shen, C.-W. Ju, J. Yi, L. Xiong, G. Ling, D. Alhmoud, H. Guan, et al., NeurIPS’2024 AI4Mat Workshop, arXiv Preprint , arXiv:2411.01578 (2024) . 80J. M. Bowman, C. Qu, R. Conte, A. Nandi, P . L....
-
[9]
6906–6919
pp. 6906–6919. 86G. Liu, Y . Shang, Y . Y ao, and R. Kompella, in2023 IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops (CVPRW) (2023) pp. 3368–3375. 87L. Y . Pratt, inAdvances in Neural Information Processing Systems, V ol. 5, edited by S. Hanson, J. Cowan, and C. Giles (Morgan-Kaufmann,
2023
-
[10]
Distilling the Knowledge in a Neural Network
pp. 3320–3328. 89J. S. Smith, B. T. Nebgen, R. Zubatyuk, N. Lubbers, C. Devereux, K. Barros, S. Tretiak, O. Isayev, and A. E. Roitberg, Nature Communications 10, 2903 (2019). 90W. Hu, B. Liu, J. Gomes, M. Zitnik, P . Liang, V . Pande, and J. Leskovec, in International Conference on Learning Representations (Curran Associates, Inc., 2020). 91G. Hinton, O. ...
work page internal anchor Pith review Pith/arXiv arXiv 2019
-
[11]
11815–11827
pp. 11815–11827. 93Y . Liu, L. Wang, M. Liu, Y . Lin, X. Zhang, B. Oztekin, and S. Ji, in 10th International Confer- ence on Learning Representations (Curran Associates, Inc., 2022). 94J. Gasteiger, F. Becker, and S. Günnemann, in Advances in Neural Information Processing Systems, V ol. 34, edited by M. Ranzato, A. Beygelzimer, Y . Dauphin, P . Liang, and...
2022
-
[12]
6790–6802
pp. 6790–6802. 95S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P . Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt, and B. Kozinsky, Nature Communications 13, 2453 (2022). 96Y . Bengio, J. Louradour, R. Collobert, and J. Weston, in Proceedings of the 26th Annual Inter- national Conference on Machine Learning , ICML ’09 (Association for Computing Machiner...
2022
-
[13]
97G. Y . Gor, S. Tapio, A. V . Domanskaya, M. Räsänen, A. V . Nemukhin, and L. Khriachtchev, Chemical Physics Letters 517, 9 (2011) . 98N. Zhang, X. Ruan, Y . Song, Z. Liu, and G. He, Journal of Molecular Liquids 221, 942 (2016). 99A. Romero, N. Ballas, S. E. Kahou, A. Chassang, C. Gatta, and Y . Bengio, in International Conference on Learning Representat...
2011
-
[14]
141Y . Mao, P . R. Horn, and M. Head-Gordon, Physical Chemistry Chemical Physics 19, 5944 (2017). 142A. Reuther, J. Kepner, C. Byun, S. Samsi, W. Arcand, D. Bestor, B. Bergeron, V . Gadepally, M. Houle, M. Hubbell, M. Jones, A. Klein, L. Milechin, J. Mullen, A. Prout, A. Rosa, C. Y ee, and P . Michaleas, in 2018 IEEE High Performance extreme Computing Con...
2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.