Recognition: unknown
Protein-Ligand Free Energy Perturbation on Quantum Hardware
Pith reviewed 2026-05-10 17:16 UTC · model grok-4.3
The pith
A quantum hardware method for protein-ligand free energy perturbation produces binding differences in reasonable agreement with experiment.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
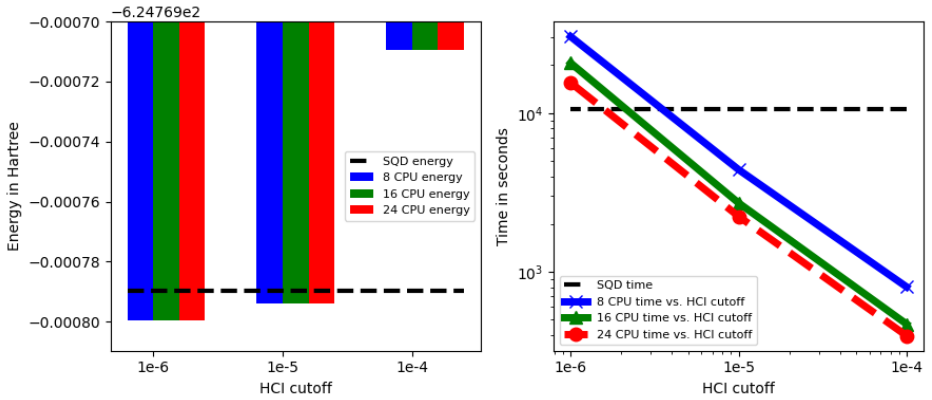
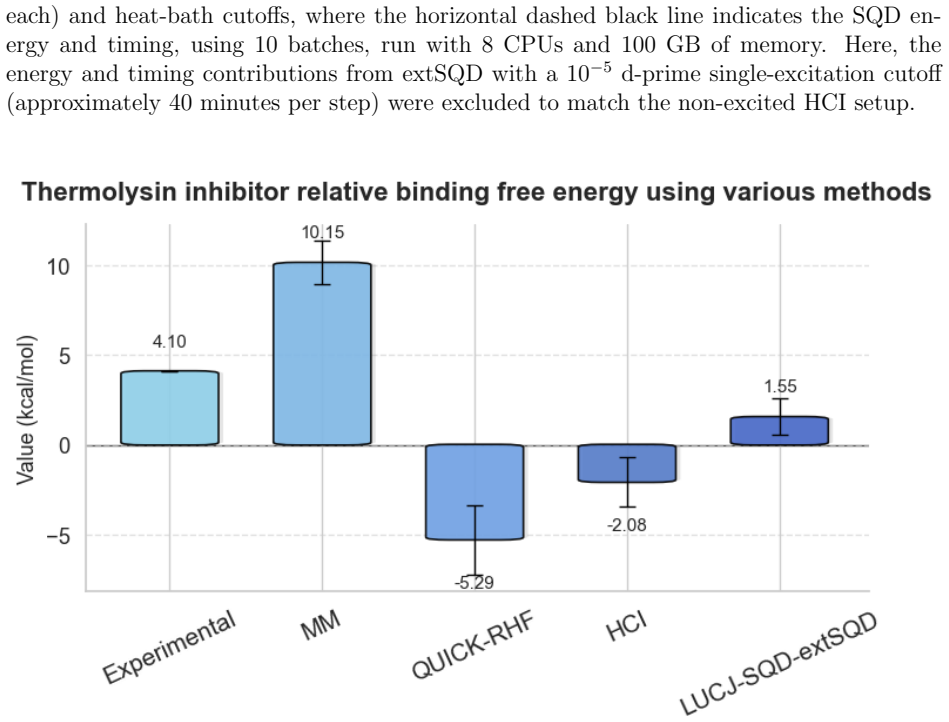
We present a QM/MM FEP method that uses AMBER with ff19SB and GAFF2 for the molecular mechanics portion of the protein and unperturbed ligand, QUICK for Hartree-Fock in the quantum region, and HCI as a classical benchmark. The same interface allows the quantum region to be treated on hardware with the LUCJ ansatz followed by SQD and extSQD post-processing. For thermolysin inhibitors the LUCJ-SQD/extSQD FEP results agree reasonably with experiment, lie closer to the experimental values than the HCI results, and require comparable execution time, indicating potential utility in the NISQ era.
What carries the argument
The Local Unitary Cluster Jastrow (LUCJ) variational ansatz executed on quantum hardware, followed by sample-based diagonalization (SQD) and extended-SQD post-processing, which supplies the quantum mechanical energies needed for the FEP difference in the QM/MM bookending scheme.
If this is right
- Protein-ligand binding free energies can be computed by replacing force-field descriptions in the quantum region with explicit quantum mechanical calculations on hardware.
- The LUCJ-SQD approach produces free-energy differences closer to experimental values than the classical HCI benchmark for the tested inhibitors.
- Execution time on quantum hardware remains comparable to classical high-level FEP, supporting practical use on NISQ devices.
- The QM/MM bookending scheme successfully connects the quantum and classical regions for these ligand systems without introducing dominant error.
Where Pith is reading between the lines
- The same hardware workflow could be applied to other protein targets to test whether quantum effects improve binding predictions in cases where force fields are known to fail.
- Further refinement of the post-processing step could reduce the number of shots needed, making larger ligands or longer alchemical transformations feasible.
- The hybrid method provides a concrete route to incorporate electronic-structure accuracy into existing FEP pipelines used in pharmaceutical screening.
Load-bearing premise
The quantum-computed energies are accurate enough that the resulting free-energy difference reflects the true binding change rather than noise or approximation error in the chosen ligand moiety.
What would settle it
Repeating the FEP calculation for the same thermolysin inhibitor pairs with a higher-accuracy classical reference method and finding that the LUCJ-SQD values deviate from both the reference and experiment by more than the reported agreement would falsify the claim that the hardware results are meaningful.
Figures



read the original abstract
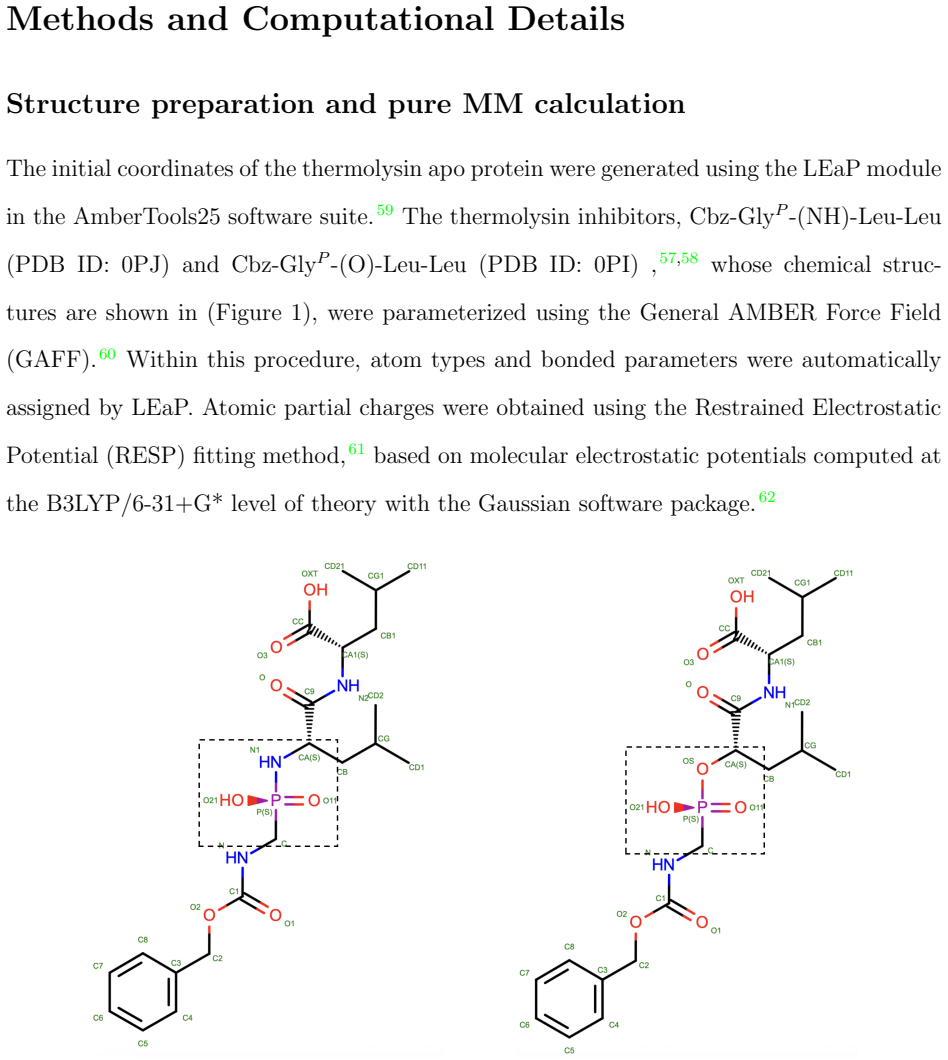
The use of free energy perturbation (FEP) methods to study protein-ligand complexes is one of the most important tools in structure-based drug design. Because FEP methods typically rely on force fields, they may suffer from force field parameter-related issues. Herein, we present a quantum mechanics/molecular mechanics (QM/MM) hybrid method to overcome deficiencies in force-field models by using QM bookending approaches on both classical and quantum hardware. In the MM part of this QM/MM FEP method, AMBER is used to simulate the protein receptor and the unperturbed moiety of the ligand, with the ff19SB and GAFF2 force fields. In the QM part, QUICK was used to conduct Hartree-Fock (HF) calculations, followed by heat-bath configuration interaction (HCI) as a benchmark on classical devices. To enable the HCI function in QUICK, we developed a Python-based interface to execute HCI from IBM's qiskit-addon-dice-solver. Moreover, the same interface also enabled this work to execute QM/MM FEP calculations on quantum hardware using the Local Unitary Cluster Jastrow (LUCJ) ansatz, followed by sample-based diagonalization (SQD) and extended-SQD (extSQD) post-processing. Using a series of thermolysis inhibitors as an example, we find reasonable agreement with experiment between the classical HCI method and the LUCJ-SQD/extSQD method, with the latter yielding a result closer to the experimental value. The execution time between the HCI-based FEP method and the LUCJ-SQD/extSQD-based FEP method is also comparable, indicating a high potential for utility in the noisy intermediate-scale quantum (NISQ) era.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a QM/MM free energy perturbation (FEP) method for protein-ligand binding free energies that combines classical MM simulations (AMBER with ff19SB/GAFF2) for the protein and unperturbed ligand moiety with a QM treatment of the perturbed moiety. The QM step uses Hartree-Fock in QUICK, benchmarked classically via heat-bath configuration interaction (HCI) through a new Python interface to qiskit-addon-dice-solver, and on quantum hardware via the Local Unitary Cluster Jastrow (LUCJ) ansatz followed by sample-based diagonalization (SQD) and extended-SQD post-processing. Applied to a series of thermolysis inhibitors, the work claims reasonable agreement with experiment for both the classical HCI and quantum LUCJ-SQD/extSQD routes, with the quantum route closer to the experimental value and with comparable execution times, indicating potential NISQ utility.
Significance. If the numerical results and error controls substantiate the claims, the work would provide a concrete demonstration that NISQ hardware can deliver QM energies sufficiently accurate for FEP differences in a drug-design context, with runtimes competitive to a classical HCI benchmark. The development of the QUICK interface for both classical HCI and quantum LUCJ-SQD/extSQD is a practical contribution that could be reused.
major comments (1)
- [Abstract] Abstract (and results section): the central claim that the LUCJ-SQD/extSQD method produces a result 'closer to the experimental value' than classical HCI rests on an assertion of 'reasonable agreement' without any reported ΔΔG values, experimental reference numbers, statistical uncertainties, number of alchemical states, QM-region atom counts, or noise-mitigation protocols. This directly prevents evaluation of whether the post-processed quantum energies are accurate enough for the FEP difference to be meaningful, as required by the weakest assumption.
Simulated Author's Rebuttal
We thank the referee for the careful reading of our manuscript and for the constructive feedback. We have addressed the major comment below and will revise the manuscript accordingly to improve clarity and completeness.
read point-by-point responses
-
Referee: [Abstract] Abstract (and results section): the central claim that the LUCJ-SQD/extSQD method produces a result 'closer to the experimental value' than classical HCI rests on an assertion of 'reasonable agreement' without any reported ΔΔG values, experimental reference numbers, statistical uncertainties, number of alchemical states, QM-region atom counts, or noise-mitigation protocols. This directly prevents evaluation of whether the post-processed quantum energies are accurate enough for the FEP difference to be meaningful, as required by the weakest assumption.
Authors: We agree that the abstract would be strengthened by including the specific quantitative details supporting the central claim. The results section of the manuscript does report the computed ΔΔG values for the thermolysin inhibitors (both HCI and LUCJ-SQD/extSQD routes), the corresponding experimental reference values, the number of alchemical states, and the QM-region sizes. However, to make these elements immediately accessible in the abstract and to explicitly address statistical uncertainties and noise-mitigation protocols, we will revise the abstract to incorporate the key numerical results (e.g., the binding free-energy differences and their deviations from experiment) along with a concise statement of the alchemical protocol and post-processing steps. This revision will allow readers to directly evaluate the accuracy of the FEP differences without needing to consult the full text first. revision: yes
Circularity Check
No significant circularity; derivation is self-contained against external benchmarks
full rationale
The paper applies a QM/MM FEP workflow (AMBER MM with QUICK/HCI or LUCJ-SQD/extSQD QM) to thermolysis inhibitors and reports numerical agreement with independent experimental ΔΔG values plus a separate classical HCI benchmark. No equations, ansatzes, or post-processing steps are shown to reduce by construction to parameters fitted on the same target data; the LUCJ ansatz and SQD/extSQD are presented as imported methods whose accuracy is tested rather than assumed. Self-citations, if present, are not load-bearing for the central claim, which rests on external experimental comparison. This is the normal non-circular outcome for a computational application paper.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption QM/MM boundary and bookending corrections introduce negligible systematic error for the ligand moiety studied
- domain assumption The LUCJ ansatz plus SQD/extSQD recovers ground-state energies sufficiently close to the exact HCI values for FEP purposes
Reference graph
Works this paper leans on
-
[1]
K.; Greenwood, J
(1) Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M. K.; Greenwood, J. et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free- 16 Energy Calculation Protocol and Force Field.Journal of the American Chemical Society 2015,137, 2695–27...
2015
-
[2]
(3) Miller, B. R. I.; McGee, T. D. J.; Swails, J. M.; Homeyer, N.; Gohlke, H.; Roitberg, A. E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations.Journal of Chemical Theory and Computation2012,8, 3314–3321, PMID: 26605738. (4) Greenidge, P. A.; Kramer, C.; Mozziconacci, J.-C.; Wolf, R. M. MM/GBSA Binding Energy Prediction on the PDBbin...
2017
-
[3]
arXiv preprint arXiv:2411.09861 , year=
(38) Hudson, P. S.; Woodcock, H. L.; Boresch, S. Use of Nonequilibrium Work Methods to Compute Free Energy Differences Between Molecular Mechanical and Quantum Me- chanical Representations of Molecular Systems.The Journal of Physical Chemistry Letters2015,6, 4850–4856, PMID: 26539729. (39) Giese, T. J.; York, D. M. Development of a Robust Indirect Approac...
-
[4]
Quantum-centric algorithm for sample-based krylov diagonalization
(53) Yu, J.; Moreno, J. R.; Iosue, J. T.; Bertels, L.; Claudino, D.; Fuller, B.; Groszkowski, P.; Humble, T. S.; Jurcevic, P.; Kirby, W. et al. Quantum-Centric Algorithm for Sample- Based Krylov Diagonalization.arXiv preprint arXiv:2501.097022025, Submitted on 16 Jan 2025 (v1), last revised 24 Jan 2025 (this version, v2). (54) Barison, S.; Robledo Moreno,...
-
[5]
(58) Merz, J., Kenneth M.; Kollman, P. A. Free energy perturbation simulations of the inhi- bition of thermolysin: prediction of the free energy of binding of a new inhibitor.Journal of the American Chemical Society1989,111, 5649–5658, doi: 10.1021/ja00197a022. (59) Case, D. A.; Cerutti, D. S.; Cruzeiro, V. W. D.; Darden, T. A.; Duke, R. E.; Ghaz- imirsae...
-
[6]
(69) Javadi-Abhari, A.; Treinish, M.; Krsulich, K.; Wood, C. J.; Lishman, J.; Gacon, J.; Martiel, S.; Nation, P. D.; Bishop, L. S.; Cross, A. W. Quantum computing with Qiskit. arXiv preprint arXiv:2405.088102024, Available athttps://arxiv.org/abs/2405. 08810. (70) Kaliakin, D.; Shajan, A.; Moreno, J. R.; Li, Z.; Mitra, A.; Motta, M.; Johnson, C.; Saki, A....
work page internal anchor Pith review arXiv
-
[7]
arXiv preprint arXiv:2503.10923 , year =
25 (72) Sayfutyarova, E. R.; Sun, Q.; Chan, G. K.-L.; Knizia, G. Automated Construction of Molecular Active Spaces from Atomic Valence Orbitals.Journal of Chemical Theory and Computation2017,13, 4063–4078. (73) Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S. PySCF: the Pytho...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.