Recognition: unknown
CovAngelo: A hybrid quantum-classical computing platform for accurate and scalable drug discovery
Pith reviewed 2026-05-10 16:15 UTC · model grok-4.3
The pith
CovAngelo computes full reaction energy profiles for covalent drug binding using a quantum-enhanced multiscale embedding model at lower cost than standard approaches.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
CovAngelo implements a new QM/QM/MM multiscale embedding model that integrates quantum-information metrics to produce entanglement-consistent orbitals, allowing the platform to calculate complete reaction energy profiles and barriers for covalent ligand-protein binding at reduced computational expense compared to existing methods while supporting multiple classical and quantum backends.
What carries the argument
The quantum-information-enhanced density matrix embedding theory that generates entanglement-consistent orbitals inside a multiscale QM/QM/MM framework with explicit solvent.
If this is right
- The platform produces full reaction energy profiles and barriers for covalent docking examples such as zanubrutinib to kinase.
- It achieves potential computational speedups of up to 20x relative to conventional methods.
- The approach scales on GPU clusters and cloud CPU systems while integrating with quantum devices of up to 20 qubits.
- Resource estimates support future use on fault-tolerant quantum hardware.
- The platform enables construction of reaction networks in chemical metric space for broader ligand screening.
Where Pith is reading between the lines
- The same embedding strategy could apply to other covalent inhibitor systems beyond the kinase example shown.
- Widespread adoption might shift drug discovery toward routine use of first-principles barrier data instead of empirical scoring functions.
- As quantum hardware matures, the hybrid design could handle larger solvated protein systems that remain intractable today.
- Reaction network construction opens the door to systematic mapping of reactive pathways across chemical libraries.
Load-bearing premise
The entanglement-consistent orbitals accurately describe the strongly correlated electronic structure at the covalent binding site without any post-hoc adjustment or experimental fitting.
What would settle it
An experimental measurement of the activation energy barrier for zanubrutinib covalent binding to Bruton's tyrosine kinase that deviates substantially from the value computed by the platform.
Figures




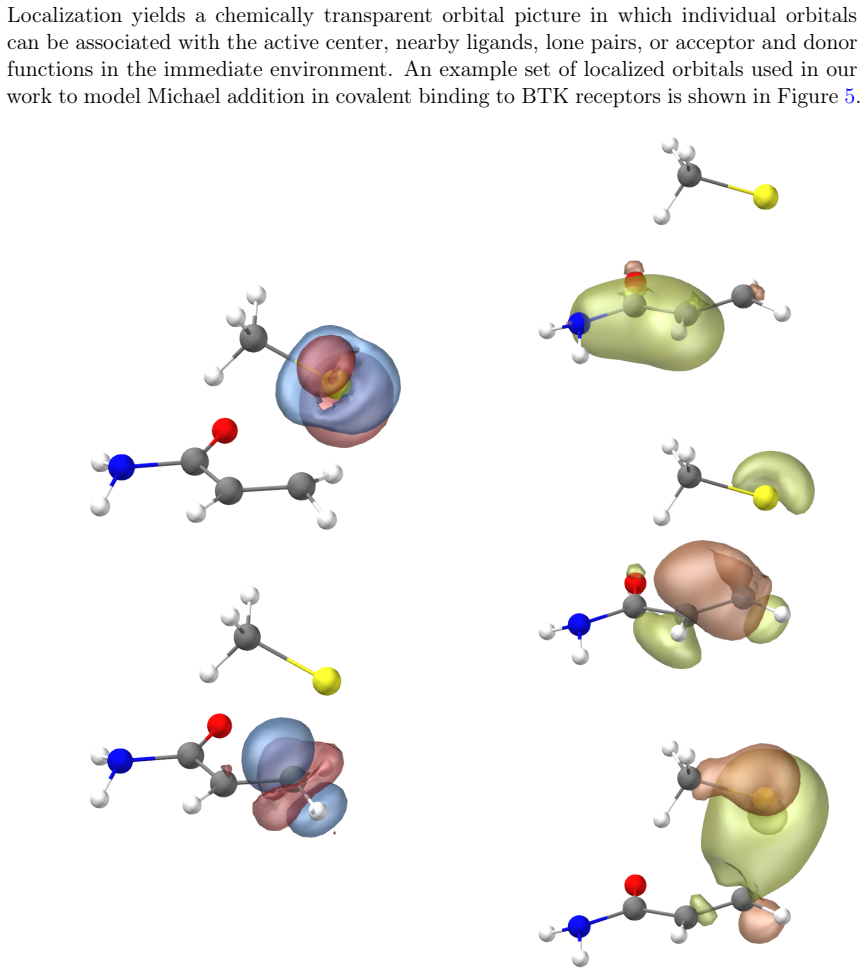


















read the original abstract
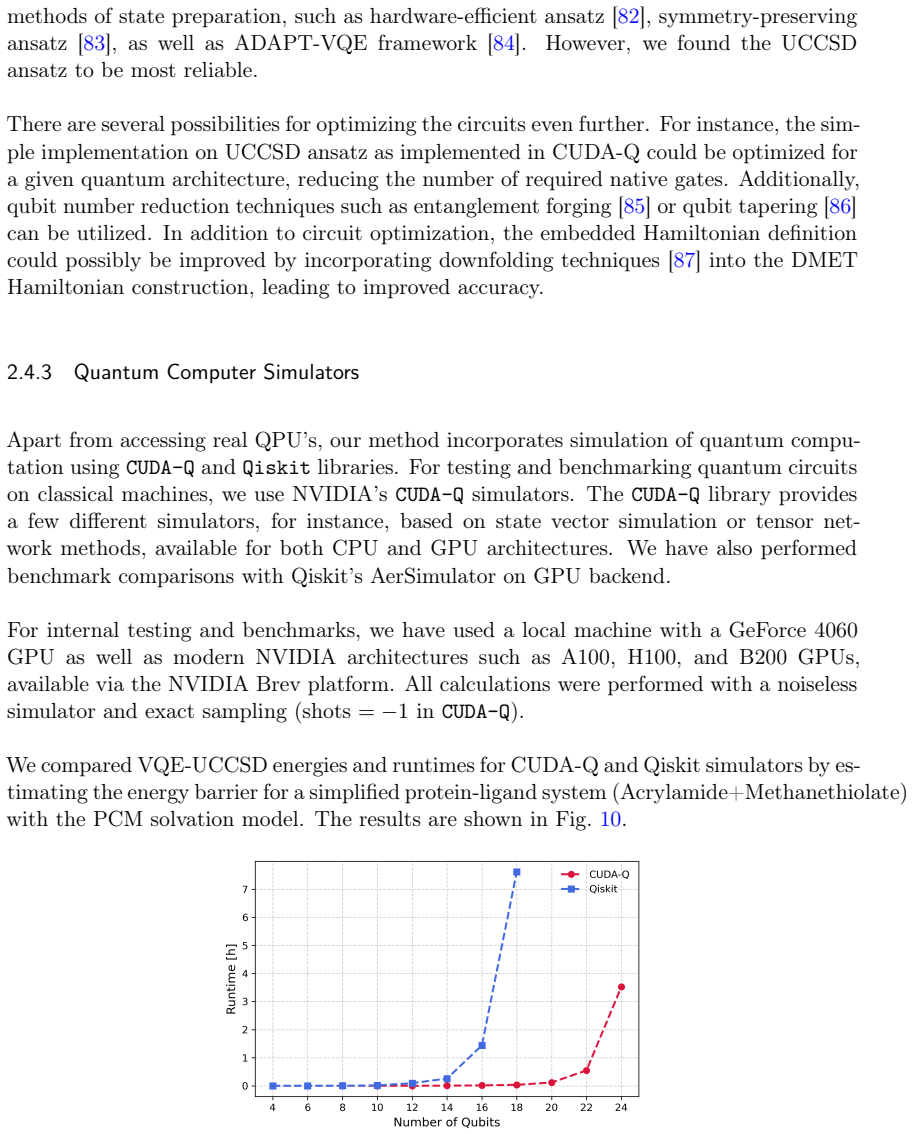
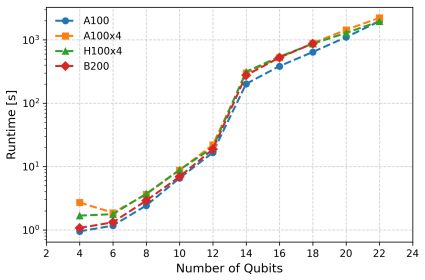
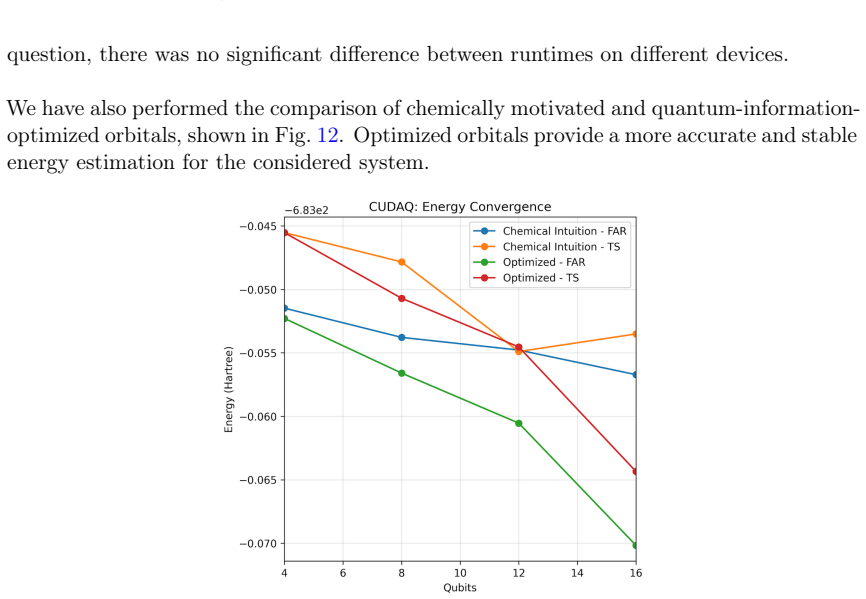
We present a computational platform for modeling chemical reactions in complex molecular environments, focused on ligand-protein binding in drug discovery. The platform implements our new quantum-in-quantum-in-classical (QM/QM/MM) multiscale embedding model that integrates molecular dynamics with a quantum-information-enhanced density matrix embedding theory and quantum chemistry solvers, including explicit solvent. Quantum-information metrics are utilized to generate entanglement-consistent orbitals, enabling a high-accuracy description of strongly correlated regions. The framework supports multiple computational backends, including multi-CPU, NVIDIA multi-GPU architectures, and quantum hardware (IQM, IonQ, IBM) integrated under CUDA-Q, and is designed for compatibility with future fault-tolerant quantum systems. The new platform's capabilities are demonstrated by modeling covalent docking of zanubrutinib to Bruton's tyrosine kinase via a Michael addition mechanism, computing the full reaction energy profiles and energy barriers at a reduced computational cost relative to existing methods. As a 2nd-generation anticancer agent, zanubrutinib serves as a proof of concept for covalent inhibitor discovery. Accurate first-principles reaction barrier estimations provided by our method can contribute to reducing false positive and negative rates in drug discovery pipelines. Scalability is validated through benchmarks on GPU clusters, cloud-based CPU infrastructures. We demonstrate integration with quantum devices (up to 20 qubits), alongside resource estimates for fault-tolerant quantum computing, indicating potential speedups of up to 20x. Beyond single reactions, the platform supports the construction of reaction networks in chemical metric space, facilitating ligand screening and systematic exploration of reactive pathways.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces CovAngelo, a hybrid quantum-classical platform implementing a QM/QM/MM multiscale embedding model that combines molecular dynamics, quantum-information-enhanced density matrix embedding theory (DMET) using entanglement-consistent orbitals, and quantum chemistry solvers with explicit solvent. It demonstrates the approach on the covalent Michael addition of zanubrutinib to BTK, claiming computation of full reaction energy profiles and barriers at reduced cost (with up to 20x speedup), integration with quantum hardware (up to 20 qubits), and potential to reduce false positives/negatives in drug discovery pipelines via accurate first-principles barriers.
Significance. If the accuracy and cost claims are substantiated, the platform could meaningfully advance computational drug discovery by enabling scalable, first-principles modeling of covalent binding reactions in complex environments, supporting reaction network construction for ligand screening. The hybrid quantum-classical design and explicit support for near-term and fault-tolerant quantum backends represent a forward-looking contribution, though the current demonstration provides no quantitative validation to assess these benefits.
major comments (3)
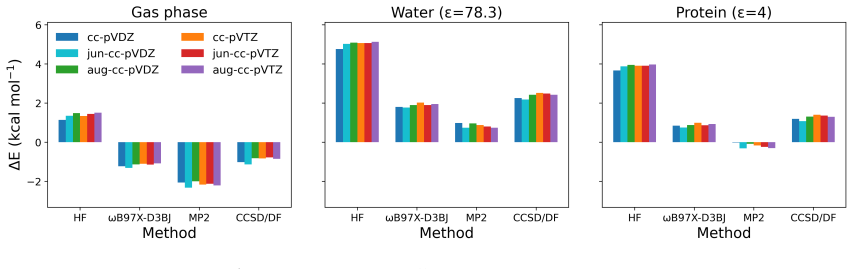
- [Abstract / Demonstration] Abstract and demonstration section: The central claims of 'accurate first-principles reaction barrier estimations' and 'reduced computational cost relative to existing methods' (including the 20x speedup figure) are asserted without any numerical barrier heights, error bars, comparison tables, or validation against experimental kinetics or gold-standard references such as CCSD(T)/CBS on the same active-site model.
- [Methods / QM/QM/MM embedding] Embedding model description: The assertion that quantum-information-enhanced DMET with entanglement-consistent orbitals 'accurately captures the strongly correlated electronic structure of the covalent binding site' lacks any benchmark data or comparison to higher-level methods, rendering the accuracy premise for the zanubrutinib-BTK profile untestable.
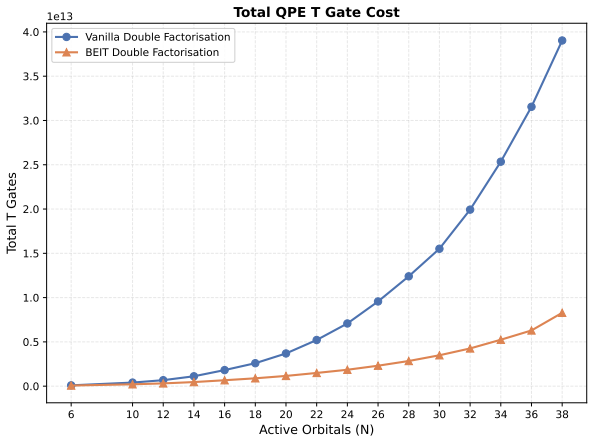
- [Results / Benchmarks] Results and benchmarks: Resource estimates for quantum integration (up to 20 qubits) and scalability on GPU/CPU clusters are mentioned without accompanying timing data, scaling plots, or direct comparisons to classical baselines for the reaction profile computation.
minor comments (2)
- [Abstract] The abstract introduces specialized terminology such as 'entanglement-consistent orbitals' and 'quantum-in-quantum-in-classical' without immediate definitions or citations to prior literature, which reduces clarity for readers unfamiliar with the subfield.
- [Methods] The manuscript would benefit from explicit statements of the active-site model size, basis sets, and active space choices used in the DMET calculations to allow reproducibility assessment.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review of our manuscript. We have addressed each major comment point by point below. We agree that additional quantitative details are required to substantiate the central claims and will revise the manuscript to incorporate the requested numerical results, benchmarks, and comparisons.
read point-by-point responses
-
Referee: [Abstract / Demonstration] Abstract and demonstration section: The central claims of 'accurate first-principles reaction barrier estimations' and 'reduced computational cost relative to existing methods' (including the 20x speedup figure) are asserted without any numerical barrier heights, error bars, comparison tables, or validation against experimental kinetics or gold-standard references such as CCSD(T)/CBS on the same active-site model.
Authors: The referee correctly notes that the abstract and demonstration section assert these claims at a high level without embedding specific numerical barrier heights, error bars, or direct comparison tables. We will revise the abstract and demonstration section to explicitly report the computed reaction barrier heights for the zanubrutinib-BTK Michael addition, associated uncertainties from the embedding procedure, and a summary table comparing to experimental kinetics where available as well as CCSD(T)/CBS results on reduced active-site models. The 20x speedup estimate, derived from resource scaling between the hybrid approach and conventional methods, will be supported with additional clarifying details on the underlying assumptions. revision: yes
-
Referee: [Methods / QM/QM/MM embedding] Embedding model description: The assertion that quantum-information-enhanced DMET with entanglement-consistent orbitals 'accurately captures the strongly correlated electronic structure of the covalent binding site' lacks any benchmark data or comparison to higher-level methods, rendering the accuracy premise for the zanubrutinib-BTK profile untestable.
Authors: We agree that the methods section would be strengthened by explicit benchmark data. The quantum-information metrics are used to select entanglement-consistent orbitals for the active space, but direct validation against higher-level methods is not currently included. In the revised manuscript we will add a dedicated benchmark subsection comparing DMET embedding energies and correlation contributions to methods such as CASPT2 and DMRG on model fragments representative of the covalent binding site, thereby providing testable support for the accuracy premise applied to the zanubrutinib-BTK profile. revision: yes
-
Referee: [Results / Benchmarks] Results and benchmarks: Resource estimates for quantum integration (up to 20 qubits) and scalability on GPU/CPU clusters are mentioned without accompanying timing data, scaling plots, or direct comparisons to classical baselines for the reaction profile computation.
Authors: The referee is correct that the results section mentions resource estimates and scalability without accompanying timing data or scaling plots. We will revise the results and benchmarks section to include wall-clock timing measurements, scaling plots versus number of processors or system size for the GPU and CPU implementations, and direct cost comparisons to standard classical QM/MM baselines for the full reaction profile computation. These additions will make the claimed reductions in computational cost and the 20-qubit quantum integration estimates quantitatively verifiable. revision: yes
Circularity Check
No significant circularity; claims rest on new model implementation without self-referential reductions.
full rationale
The manuscript describes a new QM/QM/MM embedding platform using quantum-information-enhanced DMET and entanglement-consistent orbitals, demonstrated on the zanubrutinib-BTK reaction profile. No equations, parameter fittings, or derivations appear in the provided text that would reduce any 'prediction' or barrier computation to an input by construction. Central accuracy and speedup assertions are framed as outcomes of the hybrid solver integration and hardware benchmarks rather than tautological redefinitions or self-citation chains. The absence of load-bearing self-referential steps makes the derivation chain self-contained against external validation needs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Quantum-information-enhanced density matrix embedding theory accurately describes strongly correlated regions in ligand-protein binding when supplied with entanglement-consistent orbitals.
invented entities (1)
-
entanglement-consistent orbitals
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Quantum-computing-enhanced algorithm unveils poten- tial KRAS inhibitors
M. Ghazi Vakili et al. “Quantum-computing-enhanced algorithm unveils poten- tial KRAS inhibitors”. In:Nat. Biotechnol.43.12 (2025), pp. 1954–1959.doi: 10.1038/s41587-024-02526-3
-
[2]
Innovation in the Pharmaceutical Industry: The Process of Drug Discovery and Development
E. Petrova. “Innovation in the Pharmaceutical Industry: The Process of Drug Discovery and Development”. In:Innovation and Marketing in the Pharmaceutical Industry. Springer New York, 2013, pp. 19–81.doi: 10.1007/978-1-4614-7801-0_2. 46
-
[3]
A Perspective on Covalent Inhibitors: Research and Develop- ment Trends of Warheads and Targets
H. Chen et al. “A Perspective on Covalent Inhibitors: Research and Develop- ment Trends of Warheads and Targets”. In:ChemRxiv2023.1106 (2023).doi: 10.26434/chemrxiv-2023-wf79n-v2
-
[4]
Structure and dynamics in drug discovery
H. Wei and J. A. McCammon. “Structure and dynamics in drug discovery”. In:npj Drug Discov.1 (2024), p. 1.doi: 10.1038/s44386-024-00001-2
-
[5]
Drug design on quantum computers
R. Santagati et al. “Drug design on quantum computers”. In:Nat. Phys.20.4 (2024), pp. 549–557.doi: 10.1038/s41567-024-02411-5
-
[6]
Ross, Chao Lu, Guido Scarabelli, Steven K
G. A. Ross et al. “The maximal and current accuracy of rigorous protein–ligand binding free energy calculations”. In:Communications Chemistry6.1 (2023), pp. –. doi: 10.1038/s42004-023-01019-9
-
[7]
Breaking the Molecular Dynamics Timescale Barrier Using a Wafer-Scale System
K. Santos et al. “Breaking the Molecular Dynamics Timescale Barrier Using a Wafer-Scale System”. In:arXiv preprint arXiv:2405.07898(2024).url: https: //arxiv.org/abs/2405.07898
-
[8]
Breaking the Mold: Overcoming the Time Constraints of Molecular Dynamics on General-Purpose Hardware
D. Perez et al. “Breaking the Mold: Overcoming the Time Constraints of Molecular Dynamics on General-Purpose Hardware”. In:arXiv preprint arXiv:2411.10532 (2024).url:https://arxiv.org/abs/2411.10532
-
[9]
Beyond Exascale: Dataflow Domain Translation on a Cerebras Cluster
T. Oppelstrup et al. “Beyond Exascale: Dataflow Domain Translation on a Cerebras Cluster”. In:arXiv preprint arXiv:2511.11542(2025).url: https://arxiv.org/ abs/2511.11542
-
[10]
Version accessed 2026
NVIDIA Corporation.CUDA-Q: NVIDIA Platform for Hybrid Quantum-Classical Computing. Version accessed 2026. 2024.url:https://github.com/NVIDIA/cuda- quantum
2026
-
[11]
K. Deka and E. Zak. “Simultaneously Optimizing Symmetry Shifts and Tensor Factorizations for Cost-Efficient Fault-Tolerant Quantum Simulations of Electronic Hamiltonians”. In:J. Chem. Theory Comput.21.9 (2025), pp. 4458–4465.doi: 10.1021/acs.jctc.4c01722
-
[12]
Data-Driven Strategies for Accelerated Materials Design
R. Pollice et al. “Data-Driven Strategies for Accelerated Materials Design”. In:Acc. Chem. Res.54.4 (2021), pp. 849–860.doi: 10.1021/acs.accounts.0c00785
-
[13]
Predicting transcriptional outcomes of novel multigene perturba- tions with GEARS
A. Zhavoronkov et al. “Deep learning enables rapid identification of potent DDR1 ki- naseinhibitors”.In:Nat. Biotechnol.37.9(2019),pp.1038–1040.doi:10.1038/s41587- 019-0224-x
-
[14]
Tartarus: A Benchmarking Platform for Realistic And Practical Inverse Molecular Design
A. Nigam et al. “Tartarus: A Benchmarking Platform for Realistic And Practical Inverse Molecular Design”. In:Advances in Neural Information Processing Systems. Ed. by A. Oh et al. Vol. 36. Curran Associates, Inc., 2023, pp. 3263–3306.url: https://proceedings.neurips.cc/paper_files/paper/2023/file/09f8b2469 a3d1089a7c60d9ef1983271-Paper-Datasets_and_Benchmarks.pdf
2023
-
[15]
Virtual screening of chemical libraries
B. K. Shoichet. “Virtual screening of chemical libraries”. In:Nature432.7019 (2004), pp. 862–865.doi: 10.1038/nature03197
-
[16]
Advances in covalent drug discovery
L. Boike, N. J. Henning, and D. K. Nomura. “Advances in covalent drug discovery”. In:Nat. Rev. Drug Discov.21.12 (2022), pp. 881–898.doi: 10.1038/s41573-022- 00542-z
-
[17]
J. J. Goings et al. “Reliably assessing the electronic structure of cytochrome P450 on today’s classical computers and tomorrow’s quantum computers”. In:Proc. Natl. Acad. Sci. U.S.A.119.38 (2022), e2203533119.doi: 10.1073/pnas.2203533119. 47
-
[18]
Photocatalytic Water Splitting
S. Nishioka et al. “Photocatalytic water splitting”. In:Nat. Rev. Methods Primers 3.1 (2023), p. 43.doi: 10.1038/s43586-023-00226-x
-
[19]
Artificial photosynthesis directed toward organic synthesis
S. Mori et al. “Artificial photosynthesis directed toward organic synthesis”. In:Nat. Commun.16.1 (2025), p. 1797.doi: 10.1038/s41467-025-56374-z
-
[20]
A. Anda, T. Hansen, and L. De Vico. “Multireference Excitation Energies for Bacteriochlorophylls A within Light Harvesting System 2”. In:J. Chem. Theory Comput.12.3 (2016), pp. 1305–1313.doi: 10.1021/acs.jctc.5b01104
-
[21]
Z. Li et al. “The electronic complexity of the ground-state of the FeMo cofactor of nitrogenase as relevant to quantum simulations”. In:The Journal of Chemical Physics150.2 (Jan. 2019).issn: 1089-7690.doi: 10.1063/1.5063376.url: http: //dx.doi.org/10.1063/1.5063376
-
[22]
Hunting for quantum advantage in electronic structure calculations is a highly non-trivial task,
O. Legeza et al.Hunting for quantum advantage in electronic structure calculations is a highly non-trivial task. 2026.doi: 10.48550/ARXIV.2603.28648.url: https: //arxiv.org/abs/2603.28648
-
[23]
Artificial CO2 photoreduction: a review of photocatalyst design and product selectivity regulation
C. Fu et al. “Artificial CO2 photoreduction: a review of photocatalyst design and product selectivity regulation”. In:J. Mater. Chem. A12.42 (2024), pp. 28618–28657. doi: 10.1039/d4ta04600e
-
[24]
Q. Wang and C. Pornrungroj. “Artificial photosynthetic processes using carbon dioxide, water and sunlight: can they power a sustainable future?” In:Chem. Sci. 16.41 (2025), pp. 18990–19011.doi: 10.1039/d5sc03976b
-
[25]
J.LiangandM.Head-Gordon.“Gold-StandardChemicalDatabase137(GSCDB137): A Diverse Set of Accurate Energy Differences for Assessing and Developing Density Functionals”. In:J. Chem. Theory Comput.21.24 (2025), pp. 12601–12621.doi: 10.1021/acs.jctc.5c01380
-
[26]
A Practical Guide to Density Matrix Embedding Theory in Quantum Chemistry
S. Wouters et al. “A Practical Guide to Density Matrix Embedding Theory in Quantum Chemistry”. In:J. Chem. Theory Comput.12.6 (2016), pp. 2706–2719. doi: 10.1021/acs.jctc.6b00316
-
[27]
Hierarchical Quantum Embedding by Machine Learning for Large Molecular Assemblies
M. Bensberg et al. “Hierarchical Quantum Embedding by Machine Learning for Large Molecular Assemblies”. In:J. Chem. Theory Comput.21.15 (2025), pp. 7662– 7674.doi: 10.1021/acs.jctc.5c00389
-
[28]
G. Macetti and A. Genoni. “Three-Layer Multiscale Approach Based on Extremely Localized Molecular Orbitals to Investigate Enzyme Reactions”. In:J. Phys. Chem. A125.27 (2021), pp. 6013–6027.doi: 10.1021/acs.jpca.1c05040
-
[29]
Quantum Embedding Method for the Simulation of Strongly Correlated Systems on Quantum Computers
M. Rossmannek et al. “Quantum Embedding Method for the Simulation of Strongly Correlated Systems on Quantum Computers”. In:J. Phys. Chem. Lett.14.14 (2023), pp. 3491–3497.doi: 10.1021/acs.jpclett.3c00330
-
[30]
A general framework for active space embedding methods with applications in quantum computing
S. Battaglia et al. “A general framework for active space embedding methods with applications in quantum computing”. In:npj Comput. Mater.10 (2024), p. 297.doi: 10.1038/s41524-024-01477-2
-
[31]
T. M. Bickley et al. “Extending quantum computing through subspace, embed- ding and classical molecular dynamics techniques”. In:Digit. Discov.4.12 (2025), pp. 3427–3444.doi: 10.1039/d5dd00225g
-
[32]
QM/MM simulations of EFGR with afatinib reveal the role of theβ- dimethylaminomethylsubstitution
S. Ma et al. “QM/MM simulations of EFGR with afatinib reveal the role of theβ- dimethylaminomethylsubstitution”.In:bioRxiv(2024).doi:10.1101/2024.02.18.580887. 48
-
[33]
A. Shajan et al. “Toward Quantum-Centric Simulations of Extended Molecules: Sample-Based Quantum Diagonalization Enhanced with Density Matrix Embed- ding Theory”. In:J. Chem. Theory Comput.21.14 (2025), pp. 6801–6810.doi: 10.1021/acs.jctc.5c00114
-
[34]
The Journal of Chemical Physics , author =
K. Carter-Fenk and M. Head-Gordon. “Repartitioned Brillouin-Wigner perturbation theory with a size-consistent second-order correlation energy”. In:J. Chem. Phys. 158.23 (2023), p. 234108.doi: 10.1063/5.0150033
-
[35]
An efficient real space multigrid QM/MM electrostatic coupling
T. Laino et al. “An efficient real space multigrid QM/MM electrostatic coupling”. In:J. Chem. Theory Comput.1.6 (2005), pp. 1176–1184.doi: 10.1021/ct050123f
-
[36]
T. Laino et al. “An efficient linear-scaling electrostatic coupling for treating periodic boundary conditions in QM/MM simulations”. In:J. Chem. Theory Comput.2.5 (2006), pp. 1370–1378.doi: 10.1021/ct6001169
-
[37]
Chem Shell – a modular software package for QM/MM simulations
S. Metz et al. “Chem Shell – a modular software package for QM/MM simulations”. In:WIREs Comput. Mol. Sci.4.2 (2014), pp. 101–110.doi: 10.1002/wcms.1163
-
[38]
LICHEM: A QM/MM program for simulations with multipolar and polarizable force fields
E. G. Kratz et al. “LICHEM: A QM/MM program for simulations with multipolar and polarizable force fields”. In:J. Comput. Chem.37.11 (2016), pp. 1019–1029. doi: 10.1002/jcc.24295
-
[39]
ASH:AMulti-Scale,Multi-TheoryModelingProgram
R.Bjornsson.“ASH:AMulti-Scale,Multi-TheoryModelingProgram”.In:Journal of Computational Chemistry47.8 (Mar. 2026).issn: 1096-987X.doi: 10.1002/jcc.70359. url:http://dx.doi.org/10.1002/jcc.70359
-
[40]
A hybrid quantum computing pipeline for real world drug discovery , volume=
W. Li et al. “A hybrid quantum computing pipeline for real world drug discovery”. In:Sci. Rep.14.1 (2024), p. 16942.doi: 10.1038/s41598-024-67897-8
-
[41]
Advances in molecular quantum chemistry contained in the Q-Chem 4 program package
Y. Shao et al. “Advances in molecular quantum chemistry contained in the Q-Chem 4 program package”. In:Molecular Physics113.2 (Sept. 2014), 184–215.issn: 1362- 3028.doi: 10.1080/00268976.2014.952696.url: http://dx.doi.org/10.1080/ 00268976.2014.952696
-
[42]
WIREs Computational Molecular Science , author =
F. Neese. “Software update: the ORCA program system—version 6.0”. In:WIREs Comput. Mol. Sci.15.2 (2025), e70019.doi: 10.1002/wcms.70019
-
[43]
Mechanistic aspects of thiol additions to Michael acceptors: Insights from computations
R. B. Roseli, A. B. Keto, and E. H. Krenske. “Mechanistic aspects of thiol additions to Michael acceptors: Insights from computations”. In:WIREs Comput. Mol. Sci. 13.2 (2022), e1636.doi: 10.1002/wcms.1636
-
[44]
R. Liu et al. “Quantum Descriptors for Predicting and Understanding the Structure– Activity Relationships of Michael Acceptor Warheads”. In:J. Chem. Inf. Model. 63.15 (2023), pp. 4912–4923.doi: 10.1021/acs.jcim.3c00720
-
[45]
New Means and Challenges in the Targeting of BTK
V. Nawaratne et al. “New Means and Challenges in the Targeting of BTK”. In:Clin. Cancer Res.30.11 (2024), pp. 2333–2341.doi: 10.1158/1078-0432.CCR-23-0409
-
[46]
Y. Guo et al. “Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase”. In:J. Med. Chem.62.17 (2019), pp. 7923–7940.doi: 10.1021/acs.jmedchem.9b00687
-
[47]
H. Singh et al. “Qubit-efficient quantum chemistry with the ADAPT variational quantum eigensolver and double unitary downfolding”. In:J. Chem. Theory Comput. 21.18 (2025), pp. 8799–8811.doi: 10.1021/acs.jctc.5c00896
-
[48]
Computational methods in drug discovery
G. Sliwoski et al. “Computational methods in drug discovery”. In:Pharmacol. Rev. 66.1 (2014), pp. 334–395.doi: 10.1124/pr.112.007336. 49
-
[49]
The Resurgence of Covalent Drugs
J. Singh et al. “The Resurgence of Covalent Drugs”. In:Nat. Rev. Drug Discov.10 (2011), pp. 307–317.doi: 10.1038/nrd3410
-
[50]
Covalent Docking of Large Libraries for the Discovery of Chemical Probes
N. London et al. “Covalent Docking of Large Libraries for the Discovery of Chemical Probes”. In:Nat. Chem. Biol.10.12 (2014), pp. 1066–1072.doi: 10.1038/nchem- bio.1666
-
[51]
Systematic Studies on the Protocol and Criteria for Selecting a Cova- lentDockingTool
C. Wen et al. “Systematic Studies on the Protocol and Criteria for Selecting a Cova- lentDockingTool”.In:Molecules24.11(2019),p.2183.doi:10.3390/molecules24112183
-
[52]
Comparative Evaluation of Covalent Docking Tools
A. Scarpino, G. G. Ferenczy, and G. M. Keserű. “Comparative Evaluation of Covalent Docking Tools”. In:J. Chem. Inf. Model.58.7 (2018), pp. 1441–1458.doi: 10.1021/acs.jcim.8b00228
-
[53]
Cov_DOX: A Method for Structure Prediction of Covalent Protein– LigandBindings
L. Wei et al. “Cov_DOX: A Method for Structure Prediction of Covalent Protein– LigandBindings”.In:J. Med. Chem.65.7(2022),pp.5528–5538.doi:10.1021/acs.jmedchem.1c02007
-
[54]
S. Sekaran, O. Bindech, and E. Fromager. “A unified density matrix functional construction of quantum baths in density matrix embedding theory beyond the mean-field approximation”. In:J. Chem. Phys.159.3 (2023), p. 034107.doi: 10.1063/5.0157746
-
[55]
Diagonal Operator Decomposition on Restricted Topologies via Enumeration of Quantum State Subsets
J. Tułowiecki et al. “Diagonal Operator Decomposition on Restricted Topologies via Enumeration of Quantum State Subsets”. In:arXiv:2403.02109 [quant-ph](2024)
-
[56]
M. Szczepanik and E. Zak. “Utilizing redundancies in qubit Hilbert space to reduce entangling gate counts in the unitary vibrational coupled-cluster method”. In:J. Chem. Phys.163.2 (2025), p. 024128.doi: 10.1063/5.0267596
-
[58]
M. J. Abraham et al. “GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers”. In:SoftwareX1–2 (2015), pp. 19–25.doi: 10.1016/j.softx.2015.06.001
-
[59]
M.Abrahametal.GROMACS 2026.1 Manual.2026.1.Zenodo,2026.doi:10.5281/zen- odo.18886967
-
[60]
ACPYPE - AnteChamber PYthon Parser interfacE
A. W. Sousa da Silva and W. F. Vranken. “ACPYPE - AnteChamber PYthon Parser interfacE”. In:BMC Res. Notes5.1 (2012), p. 367.doi: 10.1186/1756-0500-5-367
-
[61]
QM/MM Methods for Biomolecular Systems
H. M. Senn and W. Thiel. “QM/MM Methods for Biomolecular Systems”. In: Angew. Chem. Int. Ed.48.7 (Jan. 2009), pp. 1198–1229.issn: 1521-3773.doi: 10.1002/anie.200802019.url:http://dx.doi.org/10.1002/anie.200802019
work page doi:10.1002/anie.200802019.url:http://dx.doi.org/10.1002/anie.200802019 2009
-
[62]
J. E. Lennard-Jones. “Cohesion”. In:Proc. Phys. Soc.43.5 (1931), p. 461.doi: 10.1088/0959-5309/43/5/301
-
[63]
N. Michaud-Agrawal et al. “MDAnalysis: A toolkit for the analysis of molecular dynamics simulations”. In:J. Comput. Chem.32.10 (2011), pp. 2319–2327.doi: 10.1002/jcc.21787
-
[64]
MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations
R. J. Gowers et al. “MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations”. In:Proc. Python Sci. Conf.(2016), pp. 98–105. doi: 10.25080/Majora-629e541a-00e
-
[65]
V. Barone and M. Cossi. “Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model”. In:J. Phys. Chem. A102.11 (1998), pp. 1995–2001.doi: 10.1021/jp9716997. 50
-
[66]
A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations
M. J. Field, P. A. Bash, and M. Karplus. “A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations”. In:J. Comput. Chem.11.6 (1990), pp. 700–733.doi: https://doi.org/10.1002/jcc.540110605
-
[67]
U. C. Singh and P. A. Kollman. “A combined ab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecu- lar systems: Applications to the CH3Cl + Cl−exchange reaction and gas phase protonation of polyethers”. In:J. Comput. Chem.7.6 (1986), pp. 718–730.doi: https://doi.org/10.1002/jcc.540070604
-
[68]
QM/MM: what have we learned, where are we, and where do we go from here?
H. Lin and D. G. Truhlar. “QM/MM: what have we learned, where are we, and where do we go from here?” In:Theor. Chem. Acc.117.2 (2007), pp. 185–199.doi: 10.1007/s00214-006-0143-z
-
[69]
Characteristic bond lengths in free molecules
D. R. Lide. “Characteristic bond lengths in free molecules”. In:CRC Handbook of Chemistry and Physics. Internet Version 2005. Boca Raton, FL: CRC Press, 2005. url:http://www.hbcpnetbase.com
2005
-
[70]
B. Waszkowycz et al. “A combined quantum mechanical/molecular mechanical model of the potential energy surface of ester hydrolysis by the enzyme phos- pholipase A2”. In:J. Chem. Soc., Perkin Trans. 2(2 1991), pp. 225–231.doi: 10.1039/P29910000225
-
[71]
L.-P. Wang and C. Song. “Geometry optimization made simple with translation and rotation coordinates”. In:J. Chem. Phys.144.21 (2016), p. 214108.doi: 10.1063/1.4952956
-
[73]
S. F. Boys. “Construction of some molecular orbitals to be approximately invariant for changes from one molecule to another”. In:Rev. Mod. Phys.32.2 (1960), p. 296. doi: 10.1103/RevModPhys.32.296
-
[74]
Canonical configurational interaction procedure
J. Foster and S. F. Boys. “Canonical configurational interaction procedure”. In:Rev. Mod. Phys.32.2 (1960), p. 300.doi: 10.1103/RevModPhys.32.300
-
[75]
J. Pipek and P. G. Mezey. “A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave functions”. In:J. Chem. Phys.90.9 (1989), pp. 4916–4926.doi: 10.1063/1.456588
-
[76]
Localization of molecular orbitals: from fragments to molecule
Z. Li et al. “Localization of molecular orbitals: from fragments to molecule”. In: Acc. Chem. Res.47.9 (2014), pp. 2758–2767.doi: 10.1021/ar500082t
-
[77]
Successive Approximations by the Rayleigh-Ritz Varia- tion Method
J. K. L. MacDonald. “Successive Approximations by the Rayleigh-Ritz Varia- tion Method”. In:Physical Review43.10 (1933), 830–833.issn: 0031-899X.doi: 10.1103/physrev.43.830.url:http://dx.doi.org/10.1103/PhysRev.43.830
work page doi:10.1103/physrev.43.830.url:http://dx.doi.org/10.1103/physrev.43.830 1933
-
[78]
Completely derandomized self-adaptation in evolution strategies
N. Hansen and A. Ostermeier. “Completely Derandomized Self-Adaptation in Evolu- tionStrategies”.In:Evol. Comput.9.2(2001),pp.159–195.doi:10.1162/106365601750190398
-
[79]
Nature Communications5(1), 4213 (2014) https://doi.org/ 10.1038/ncomms5213
A. Peruzzo et al. “A variational eigenvalue solver on a photonic quantum processor”. In:Nat. Commun.5.1 (2014), p. 4213.doi: 10.1038/ncomms5213
-
[80]
C. Angeli, R. Cimiraglia, and J.-P. Malrieu. “N-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and partially contracted variants”. In:J. Chem. Phys.117.20 (2002), pp. 9138–9153.doi: 10.1063/1.1515317. 51
-
[81]
J. Finley et al. “The multi-state CASPT2 method”. In:Chem. Phys. Lett.288.2-4 (1998), pp. 299–306.doi: 10.1016/S0009-2614(98)00252-8
-
[82]
On the practical usefulness of the Hardware Efficient Ansatz
L. Leone et al. “On the practical usefulness of the Hardware Efficient Ansatz”. In: Quantum8 (2024), p. 1395.doi: 10.22331/q-2024-07-03-1395
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.