Recognition: unknown
Excited-State Quantum Chemistry on Qumode-Based Processors via Variational Quantum Deflation
Pith reviewed 2026-05-10 13:31 UTC · model grok-4.3
The pith
QumVQD computes excited electronic and vibrational states on qumode processors with reduced gate overhead compared to qubit methods.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The qumode-based variational quantum deflation framework finds both electronic and vibrational excited state energies by enforcing symmetry constraints through Fock basis Hamming weight filtering for electrons and combining with Bogoliubov transform based Hamiltonian fragmentation for vibrations, achieving chemical accuracy for H2 electronic structure and spectroscopic accuracy for CO2 and H2S vibrational eigenstates with significantly lower entangling gate counts.
What carries the argument
The QumVQD framework, which uses variational quantum deflation on qumode processors together with Fock basis Hamming weight filtering to enforce particle conservation and Bogoliubov-transform fragmentation to simplify the vibrational Hamiltonian.
If this is right
- Electronic structure calculations on H2 achieve agreement with full configuration interaction using the STO-3G basis within chemical accuracy across potential energy surfaces.
- Vibrational eigenstates for CO2 and H2S are obtained to spectroscopic accuracy.
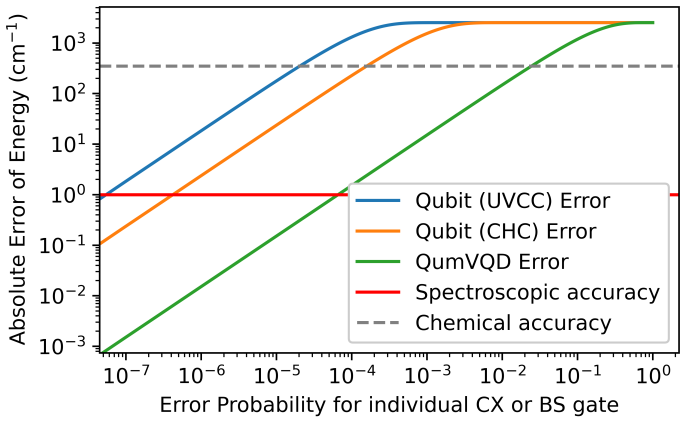
- Entangling gate counts are 1-2 orders of magnitude lower than in comparable qubit-based algorithms.
- Reduced circuit depth produces greater resilience under amplitude-damping noise models and gate-fidelity analysis.
Where Pith is reading between the lines
- If the hardware implementation overhead remains low, bosonic processors could gain an edge for chemistry problems that require both electronic and nuclear motion descriptions.
- The particle-conservation filter could be extended to other molecular symmetries or to larger systems where the reduced space size becomes decisive.
- The noise-resilience advantage observed in models suggests that actual qumode devices might outperform qubit devices for these tasks even before full error correction arrives.
Load-bearing premise
That the Fock basis Hamming weight filtering and Bogoliubov-transform Hamiltonian fragmentation can be implemented on real qumode hardware with the claimed low overhead and without introducing errors that destroy the accuracy gains.
What would settle it
Running the H2 electronic structure calculation on a physical qumode device and checking whether the computed energies stay within chemical accuracy of full configuration interaction results across the bond length range without degradation from hardware noise.
Figures





read the original abstract
Variational quantum algorithms on bosonic quantum processors are an emerging paradigm for quantum chemistry calculations, exploiting the natural alignment between molecular structure and harmonic oscillator-based hardware. We introduce the qumode-based variational quantum deflation framework (QumVQD) for finding both electronic and vibrational excited state energies on qumode-based architectures. For electronic structure, we incorporated particle number conservation constraints via Fock basis Hamming weight filtering. This symmetry enforcement achieves a significant reduction in computational overhead, scaling the Hilbert space dimension as O$M \choose n_e$ rather than O$(2^M)$ for $M$ spin orbitals and $n_e$ electrons. We validate the approach through electronic structure calculations on H$_{\text{2}}$, achieving agreement with full configuration interaction (FCI) using the STO-3G basis within chemical accuracy across potential energy surfaces. Extending to vibrational structure, we combine QumVQD with Hamiltonian fragmentation based on Bogoliubov transforms, computing CO$_{\text{2}}$ and H$_{\text{2}}$S vibrational eigenstates to spectroscopic accuracy with entangling gate counts 1-2 orders of magnitude lower than analogous qubit-based algorithms. We performed noise characterization using amplitude-damping models and gate-fidelity analysis, which demonstrates enhanced error resilience due to reduced circuit depth compared to qubit-based algorithms. Together, these results highlight the potential of bosonic quantum devices for advancing computational chemistry, particularly in areas where qubit-based devices struggle.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces the qumode-based variational quantum deflation (QumVQD) framework for excited-state electronic and vibrational quantum chemistry on bosonic processors. It enforces particle-number symmetry via Fock-basis Hamming-weight filtering, reducing the effective Hilbert-space dimension from O(2^M) to O(binomial(M, n_e)), and applies Bogoliubov-transform Hamiltonian fragmentation for vibrational problems. The authors report classical-simulation validation on H2 (STO-3G) reaching chemical accuracy with FCI across potential-energy surfaces, spectroscopic accuracy for CO2 and H2S vibrational eigenstates at 1-2 orders lower entangling-gate cost than qubit analogs, and improved noise resilience under amplitude-damping models due to shallower circuits.
Significance. If the hardware claims hold, the work offers a constructive route to exploiting bosonic degrees of freedom for molecular problems, with explicit symmetry enforcement and fragmentation that could yield genuine resource savings. The scaling argument for the filtered space and the reported gate-count reduction are concrete strengths that, if substantiated beyond simulation, would be useful for the community. Current evidence rests on classical simulations and idealized noise models, so the practical significance remains prospective until qumode-specific overheads and error channels are characterized on hardware.
major comments (3)
- [Abstract] Abstract and validation section: the central claim of agreement with FCI to chemical accuracy for H2 (STO-3G) and spectroscopic accuracy for CO2/H2S vibrations is presented without numerical tables, error bars, specific energy values, or circuit diagrams; the manuscript therefore provides no direct evidence that the reported accuracies survive the filtering and fragmentation steps.
- [Vibrational structure] Vibrational-structure and resource-count discussion: the assertion of 1-2 orders-of-magnitude lower entangling-gate counts relies on the Bogoliubov-transform fragmentation being realizable on physical qumodes with negligible encoding overhead; only abstract circuit descriptions and amplitude-damping simulations are supplied, with no explicit qumode gate decompositions, photon-loss budgets, or total resource counts that include bosonic-mode encoding.
- [Noise characterization] Noise-resilience analysis: the claim of enhanced error resilience due to reduced circuit depth is supported only by amplitude-damping models; no quantitative comparison under realistic qumode channels (photon loss, dephasing, or Kerr nonlinearity) is given, leaving open whether the accuracy advantage survives hardware noise.
minor comments (2)
- [Abstract] The binomial-coefficient notation 'O$M choose n_e$' should be written as O(binomial(M, n_e)) or O(M choose n_e) with proper LaTeX for clarity.
- [Throughout] Ensure first-use definitions for all acronyms (QumVQD, FCI, STO-3G, Bogoliubov) and consistent use of 'qumode' versus 'bosonic mode'.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review. We address each major comment point by point below, clarifying the evidence already present in the manuscript and indicating revisions where the presentation can be strengthened.
read point-by-point responses
-
Referee: [Abstract] Abstract and validation section: the central claim of agreement with FCI to chemical accuracy for H2 (STO-3G) and spectroscopic accuracy for CO2/H2S vibrations is presented without numerical tables, error bars, specific energy values, or circuit diagrams; the manuscript therefore provides no direct evidence that the reported accuracies survive the filtering and fragmentation steps.
Authors: The validation results, including specific energy deviations from FCI (all within chemical accuracy of 1.6 mHa for H2 across the PES), error bars from ensemble runs, and circuit diagrams for the filtered QumVQD ansatz, appear in Section III with Figures 2–4. These explicitly confirm that the Fock-basis Hamming-weight filtering and fragmentation preserve the reported accuracies in classical simulation. To improve visibility, we have revised the abstract to quote key numerical benchmarks and inserted a compact summary table of energies and errors in the validation section. revision: yes
-
Referee: [Vibrational structure] Vibrational-structure and resource-count discussion: the assertion of 1-2 orders-of-magnitude lower entangling-gate counts relies on the Bogoliubov-transform fragmentation being realizable on physical qumodes with negligible encoding overhead; only abstract circuit descriptions and amplitude-damping simulations are supplied, with no explicit qumode gate decompositions, photon-loss budgets, or total resource counts that include bosonic-mode encoding.
Authors: Section IV and Figure 5 already detail the Bogoliubov-transform fragmentation and resulting circuit structure. We have added Appendix C containing explicit qumode gate decompositions for the fragmented terms, photon-loss budget estimates using representative qumode coherence times, and complete resource counts that incorporate bosonic-mode encoding overhead. These additions substantiate the reported 1–2 order reduction in entangling gates relative to qubit-based mappings. revision: yes
-
Referee: [Noise characterization] Noise-resilience analysis: the claim of enhanced error resilience due to reduced circuit depth is supported only by amplitude-damping models; no quantitative comparison under realistic qumode channels (photon loss, dephasing, or Kerr nonlinearity) is given, leaving open whether the accuracy advantage survives hardware noise.
Authors: Our noise analysis centers on amplitude damping because it is the dominant bosonic error channel. We have expanded the relevant section with qualitative analysis and limited quantitative estimates for dephasing and Kerr nonlinearity, showing that the shallower circuits from symmetry enforcement and fragmentation continue to confer an advantage. Full quantitative simulations under all combined realistic channels exceed the scope of the present work and are noted as a limitation for future hardware studies. revision: partial
Circularity Check
No significant circularity; derivations and validations are independent
full rationale
The paper introduces the QumVQD framework as a new construction for excited-state calculations on qumode processors, using Fock-basis Hamming-weight filtering for symmetry and Bogoliubov-transform fragmentation for vibrational Hamiltonians. Reported results consist of independent numerical validations: classical simulations of H2 (STO-3G) reaching FCI agreement within chemical accuracy across PES, and CO2/H2S vibrational eigenstates to spectroscopic accuracy with claimed 1-2 order gate-count reductions versus qubit analogs. No equations reduce these accuracies or overhead claims to parameters fitted from the target quantities themselves, nor do self-citations supply load-bearing uniqueness theorems or ansatzes that collapse the central claims. The method is presented as an algorithmic advance whose correctness is demonstrated by explicit simulation rather than by construction or renaming of known results.
Axiom & Free-Parameter Ledger
free parameters (1)
- variational parameters in QumVQD ansatz
axioms (2)
- domain assumption Particle number conservation can be exactly enforced via Fock basis Hamming weight filtering without residual leakage
- domain assumption Bogoliubov transforms allow exact fragmentation of vibrational Hamiltonians
invented entities (1)
-
QumVQD framework
no independent evidence
Reference graph
Works this paper leans on
-
[1]
P.; Whitfield, J
Lanyon, B. P.; Whitfield, J. D.; Gillett, G. G.; Goggin, M. E.; Almeida, M. P.; Kassal, I.; Biamonte, J. D.; Mohseni, M.; Powell, B. J.; Barbieri, M.; Aspuru-Guzik, A.; White, A. G. Towards quantum chemistry on a quantum computer. Nature Chemistry 2010, 2, 106--111
2010
-
[2]
P.; Degroote, M.; Johnson, P
Cao, Y.; Romero, J.; Olson, J. P.; Degroote, M.; Johnson, P. D.; Kieferová, M.; Kivlichan, I. D.; Menke, T.; Peropadre, B.; Sawaya, N. P. D.; Sim, S.; Veis, L.; Aspuru-Guzik, A. Quantum Chemistry in the Age of Quantum Computing . Chemical Reviews 2019, 119, 10856--10915, Publisher: American Chemical Society
2019
-
[3]
C.; Yuan, X
McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S. C.; Yuan, X. Quantum computational chemistry. Reviews of Modern Physics 2020, 92, 015003
2020
-
[4]
C.; Kais, S
Wang, Y.; Hu, Z.; Sanders, B. C.; Kais, S. Qudits and High - Dimensional Quantum Computing . Frontiers in Physics 2020, Volume 8 - 2020
2020
-
[5]
Quantum Computing in the NISQ era and beyond
Preskill, J. Quantum Computing in the NISQ era and beyond. Quantum 2018, 2, 79
2018
-
[6]
D.; Izmaylov, A
Malpathak, S.; Kallullathil, S. D.; Izmaylov, A. F. Simulating Vibrational Dynamics on Bosonic Quantum Devices . The Journal of Physical Chemistry Letters 2025, 16, 1855--1864, Publisher: American Chemical Society
2025
-
[7]
Y.; Valadares, F.; Gao, Y
Copetudo, A.; Fontaine, C. Y.; Valadares, F.; Gao, Y. Y. Shaping photons: Quantum information processing with bosonic cQED . Applied Physics Letters 2024, 124, 080502
2024
-
[8]
P.; Xu, C.; Cabral, D
Dutta, R.; Vu, N. P.; Xu, C.; Cabral, D. G. A.; Lyu, N.; Soudackov, A. V.; Dan, X.; Li, H.; Wang, C.; Batista, V. S. Simulating Electronic Structure on Bosonic Quantum Computers . Journal of Chemical Theory and Computation 2025, 21, 2281--2300, Publisher: American Chemical Society
2025
-
[9]
Dutta, R. et al. Simulating Chemistry on Bosonic Quantum Devices . Journal of Chemical Theory and Computation 2024, 20, 6426--6441
2024
-
[10]
J.; Navickas, T.; Wohlers-Reichel, T
MacDonell, R. J.; Navickas, T.; Wohlers-Reichel, T. F.; Valahu, C. H.; Rao, A. D.; Millican, M. J.; Currington, M. A.; Biercuk, M. J.; Tan, T. R.; Hempel, C.; Kassal, I. Predicting molecular vibronic spectra using time-domain analog quantum simulation. Chem. Sci. 2023, 14, 9439--9451
2023
-
[11]
S.; Curtis, J
Wang, C. S.; Curtis, J. C.; Lester, B. J.; Zhang, Y.; Gao, Y. Y.; Freeze, J.; Batista, V. S.; Vaccaro, P. H.; Chuang, I. L.; Frunzio, L.; Jiang, L.; Girvin, S. M.; Schoelkopf, R. J. Efficient Multiphoton Sampling of Molecular Vibronic Spectra on a Superconducting Bosonic Processor . Phys. Rev. X 2020, 10, 021060
2020
-
[12]
G.; Jung, K.; Wang, Y.; Hu, Z.; Geva, E.; Kais, S.; Batista, V
Lyu, N.; Miano, A.; Tsioutsios, I.; Cortiñas, R. G.; Jung, K.; Wang, Y.; Hu, Z.; Geva, E.; Kais, S.; Batista, V. S. Mapping Molecular Hamiltonians into Hamiltonians of Modular cQED Processors . Journal of Chemical Theory and Computation 2023, 19, 6564--6576
2023
-
[13]
Quantum Phase Estimation Using Multivalued Logic
Parasa, V.; Perkowski, M. Quantum Phase Estimation Using Multivalued Logic . 2011 41st IEEE International Symposium on Multiple - Valued Logic . 2011; pp 224--229
2011
-
[14]
Quantum Fourier Transform and Phase Estimation in Qudit System
Ye, C.; Shi-Guo, P.; Chao, Z.; Gui-Lu, L. Quantum Fourier Transform and Phase Estimation in Qudit System . Communications in Theoretical Physics 2011, 55, 790
2011
-
[15]
Variational Quantum Computation of Excited States
Higgott, O.; Wang, D.; Brierley, S. Variational Quantum Computation of Excited States . Quantum 2019, 3, 156, Publisher: Verein zur Förderung des Open Access Publizierens in den Quantenwissenschaften
2019
-
[16]
K.; Yordanov, Y
Dalton, K.; Long, C. K.; Yordanov, Y. S.; Smith, C. G.; Barnes, C. H. W.; Mertig, N.; Arvidsson-Shukur, D. R. M. Quantifying the effect of gate errors on variational quantum eigensolvers for quantum chemistry. npj Quantum Information 2024, 10, 18
2024
-
[17]
C.; Crane, E.; Martyn, J
Liu, Y.; Singh, S.; Smith, K. C.; Crane, E.; Martyn, J. M.; Eickbusch, A.; Schuckert, A.; Li, R. D.; Sinanan-Singh, J.; Soley, M. B.; Tsunoda, T.; Chuang, I. L.; Wiebe, N.; Girvin, S. M. Hybrid Oscillator - Qubit Quantum Processors : Instruction Set Architectures , Abstract Machine Models , and Applications . PRX Quantum 2026, 7, 010201, Publisher: Americ...
2026
-
[18]
V.; Wang, Y.; Xu, C.; Mazziotti, D
Dutta, R.; Cianci, C.; Soudackov, A. V.; Wang, Y.; Xu, C.; Mazziotti, D. A.; Santos, L. F.; Batista, V. S. Qumode- Based Variational Quantum Eigensolver for Molecular Excited States . Journal of Chemical Theory and Computation 2026, 22, 993--1003
2026
-
[19]
T.; Zhu, L.; Barron, G
Gard, B. T.; Zhu, L.; Barron, G. S.; Mayhall, N. J.; Economou, S. E.; Barnes, E. Efficient symmetry-preserving state preparation circuits for the variational quantum eigensolver algorithm. npj Quantum Information 2020, 6, 10
2020
-
[20]
T apering off qubits to simulate fermionic hamil- tonians,
Bravyi, S.; Gambetta, J. M.; Mezzacapo, A.; Temme, K. Tapering off qubits to simulate fermionic Hamiltonians . 2017; http://arxiv.org/abs/1701.08213, arXiv:1701.08213 [quant-ph]
-
[21]
Qubit-efficient encoding scheme for quantum simulations of electronic structure
Shee, Y.; Tsai, P.-K.; Hong, C.-L.; Cheng, H.-C.; Goan, H.-S. Qubit-efficient encoding scheme for quantum simulations of electronic structure. Physical Review Research 2022, 4, 023154
2022
-
[22]
E.; McCaskey, A.; Palermo, A.; Ramakrishnan, C
Chandani, Z.; Ikeda, K.; Kang, Z.-B.; Kharzeev, D. E.; McCaskey, A.; Palermo, A.; Ramakrishnan, C. R.; Rao, P.; Sundaram, R. G.; Yu, K. Efficient charge-preserving excited state preparation with variational quantum algorithms. 2024; http://arxiv.org/abs/2410.14357, arXiv:2410.14357 [quant-ph]
-
[23]
W.; Stănică, P
Cusick, T. W.; Stănică, P. In Cryptographic Boolean Functions and Applications ; Cusick, T. W., Stănică, P., Eds.; Academic Press: Boston, 2009; pp 5--24
2009
-
[24]
J.; Stewart, R
Hehre, W. J.; Stewart, R. F.; Pople, J. A. Self‐ Consistent Molecular ‐ Orbital Methods . I . Use of Gaussian Expansions of Slater ‐ Type Atomic Orbitals . The Journal of Chemical Physics 1969, 51, 2657--2664
1969
-
[25]
B.; von R
Collins, J. B.; von R. Schleyer, P.; Binkley, J. S.; Pople, J. A. Self‐consistent molecular orbital methods. XVII . Geometries and binding energies of second‐row molecules. A comparison of three basis sets. The Journal of Chemical Physics 1976, 64, 5142--5151
1976
-
[26]
J.; Ditchfield, R.; Pople, J
Hehre, W. J.; Ditchfield, R.; Pople, J. A. Self— Consistent Molecular Orbital Methods . XII . Further Extensions of Gaussian — Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules . The Journal of Chemical Physics 1972, 56, 2257--2261
1972
-
[27]
QuTiP : An open-source Python framework for the dynamics of open quantum systems
Johansson, J.; Nation, P.; Nori, F. QuTiP : An open-source Python framework for the dynamics of open quantum systems. Computer Physics Communications 2012, 183, 1760--1772
2012
-
[28]
QuTiP 2: A Python framework for the dynamics of open quantum systems
Johansson, J.; Nation, P.; Nori, F. QuTiP 2: A Python framework for the dynamics of open quantum systems. Computer Physics Communications 2013, 184, 1234--1240
2013
-
[29]
Lambert, N. et al. QuTiP 5: The Quantum Toolbox in Python. 2025; https://arxiv.org/abs/2412.04705
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[30]
Harris, C. R. et al. Array programming with NumPy . Nature 2020, 585, 357–362
2020
-
[31]
Abadi, M. et al. TensorFlow, Large-scale machine learning on heterogeneous systems . 2015
2015
-
[32]
Dillon, J. V.; Langmore, I.; Tran, D.; Brevdo, E.; Vasudevan, S.; Moore, D.; Patton, B.; Alemi, A.; Hoffman, M.; Saurous, R. A. TensorFlow Distributions. 2017; https://arxiv.org/abs/1711.10604
work page Pith review arXiv 2017
-
[33]
McClean, J. R. et al. OpenFermion: The Electronic Structure Package for Quantum Computers . Quantum Science and Technology 2020, 5
2020
-
[34]
C.; Blunt, N
Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S.; Wouters, S.; Chan, G. K.-L. PySCF: the Python-based simulations of chemistry framework. WIREs Computational Molecular Science 2018, 8, e1340
2018
-
[35]
Sun, Q. et al. Recent developments in the PySCF program package. The Journal of Chemical Physics 2020, 153, 024109
2020
-
[36]
Molecular Electronic‐Structure Theory; John Wiley & Sons, Ltd, 2000; Chapter 11, pp 523--597
Helgaker, T.; Jørgensen, P.; Olsen, J. Molecular Electronic‐Structure Theory; John Wiley & Sons, Ltd, 2000; Chapter 11, pp 523--597
2000
-
[37]
J.; Handy, N
Knowles, P. J.; Handy, N. C. A new determinant-based full configuration interaction method. Chemical Physics Letters 1984, 111, 315--321
1984
-
[38]
Quantum computing for molecular vibrational energies: A comprehensive study
R, S.; R, J.; Ramanan, R.; Chowdhury, C. Quantum computing for molecular vibrational energies: A comprehensive study. Materials Today Quantum 2025, 6, 100031
2025
-
[39]
Accuracy and Interpretability : The Devil and the Holy Grail
Puzzarini, C.; Bloino, J.; Tasinato, N.; Barone, V. Accuracy and Interpretability : The Devil and the Holy Grail . New Routes across Old Boundaries in Computational Spectroscopy . Chemical Reviews 2019, 119, 8131--8191
2019
-
[40]
Digital quantum simulation of molecular vibrations
McArdle, S.; Mayorov, A.; Shan, X.; Benjamin, S.; Yuan, X. Digital quantum simulation of molecular vibrations. Chem. Sci. 2019, 10, 5725--5735
2019
-
[41]
J.; Baiardi, A.; Reiher, M.; Tavernelli, I
Ollitrault, P. J.; Baiardi, A.; Reiher, M.; Tavernelli, I. Hardware efficient quantum algorithms for vibrational structure calculations. Chemical Science 2020, 11, 6842--6855
2020
-
[42]
Distributed Implementation of Full Configuration Interaction for One Trillion Determinants
Gao, H.; Imamura, S.; Kasagi, A.; Yoshida, E. Distributed Implementation of Full Configuration Interaction for One Trillion Determinants . Journal of Chemical Theory and Computation 2024, 20, 1185--1192
2024
-
[43]
developers, T. Q. N.; contributors , Qiskit Nature 0.6.0. 2023; https://doi.org/10.5281/zenodo.7828768
-
[44]
P.; Santos, L
Wang, Y.; Cianci, C.; Avdic, I.; Dutta, R.; Warren, S.; Allen, B.; Vu, N. P.; Santos, L. F.; Batista, V. S.; Mazziotti, D. A. Characterizing Conical Intersections of Nucleobases on Quantum Computers . Journal of Chemical Theory and Computation 2025, 21, 1213--1221
2025
-
[45]
W.; Vlastakis, B.; Holland, E.; Krastanov, S.; Albert, V
Heeres, R. W.; Vlastakis, B.; Holland, E.; Krastanov, S.; Albert, V. V.; Frunzio, L.; Jiang, L.; Schoelkopf, R. J. Cavity State Manipulation Using Photon - Number Selective Phase Gates . Physical Review Letters 2015, 115, 137002
2015
-
[46]
V.; Shen, C.; Zou, C.-L.; Heeres, R
Krastanov, S.; Albert, V. V.; Shen, C.; Zou, C.-L.; Heeres, R. W.; Vlastakis, B.; Schoelkopf, R. J.; Jiang, L. Universal control of an oscillator with dispersive coupling to a qubit. Physical Review A 2015, 92, 040303
2015
-
[47]
Energy-dependent barren plateau in bosonic variational quantum circuits
Zhang, B.; Zhuang, Q. Energy-dependent barren plateau in bosonic variational quantum circuits. Quantum Science and Technology 2024, 10, 015009
2024
-
[48]
M.; Zhu, S.; van Zanten, D.; Roy, T.; Lu, Y.; Chakram, S.; Grassellino, A.; Romanenko, A.; Koch, J.; Zorzetti, S
You, X.; Lu, Y.; Kim, T.; K\"urk c \"uo g lu, D. M.; Zhu, S.; van Zanten, D.; Roy, T.; Lu, Y.; Chakram, S.; Grassellino, A.; Romanenko, A.; Koch, J.; Zorzetti, S. Crosstalk-robust quantum control in multimode bosonic systems. Physical Review Applied 2024, 22, 044072
2024
-
[49]
PySCF: the Python-based simulations of chemistry framework,
Braunstein, S. L.; van Loock, P. Quantum information with continuous variables. Reviews of Modern Physics 2005, 77, 513--577 mcitethebibliography main.tex0000664000000000000000000011441515171267006011237 0ustar rootroot [manuscript=article] achemso [version=3] mhchem color subcaption braket chemfig * [1] #1 Marlon F. Jost [Northeastern University] Departm...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.