Recognition: unknown
NEPMaker: Active learning of neuroevolution machine learning potential for large cells
Pith reviewed 2026-05-10 12:13 UTC · model grok-4.3
The pith
Active learning embeds extrapolative atomic environments from large simulations into locally periodic structures to build reliable machine learning potentials.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The framework identifies extrapolative atomic environments on-the-fly during large-scale simulations and embeds them into locally periodic structures where boundary atoms are optimized to remain close to the training distribution. This strategy enables large-scale simulations to directly contribute to dataset construction, significantly reducing extrapolation errors while improving model robustness and transferability.
What carries the argument
The on-the-fly identification and embedding of extrapolative atomic environments into locally periodic structures with optimized boundary atoms, which carries the argument by allowing large simulations to expand the training set safely.
Load-bearing premise
That embedding extrapolative environments into locally periodic structures with optimized boundary atoms will reliably reduce extrapolation errors without introducing new biases or artifacts in the training data.
What would settle it
Apply the trained potential to a new large-cell simulation containing the previously extrapolative environments and check whether force or energy errors stay low compared with direct first-principles calculations; persistent high errors would falsify the claim.
Figures

read the original abstract
Machine learning potentials (MLPs) achieve near first-principles accuracy but often fail for atomic environments outside the training distribution. Active learning can mitigate this limitation; however, its application to large-scale simulations is hindered by the prohibitive cost of labeling entire configurations. Here, we develop a D-optimality-driven active learning framework for the neuroevolution potential (NEP) implemented within the GPUMD package, named NEPMaker. Extrapolative atomic environments are identified on-the-fly and embedded into locally periodic structures, where boundary atoms are optimized to remain close to the training distribution. This strategy enables large-scale simulations to directly contribute to dataset construction, significantly reducing extrapolation errors while improving model robustness and transferability. The proposed framework provides a scalable route for constructing reliable machine learning potentials in complex materials systems, including those involving defects, interfaces, and phase transitions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces NEPMaker, a D-optimality-driven active learning framework for neuroevolution potentials (NEPs) within the GPUMD package. It identifies extrapolative atomic environments on-the-fly during large-cell molecular dynamics simulations, embeds them into locally periodic supercells, and optimizes boundary atoms to remain near the training distribution. This enables large-scale configurations to contribute directly to training data construction, with the goal of reducing extrapolation errors and improving robustness for systems involving defects, interfaces, and phase transitions.
Significance. If the embedding and optimization steps preserve local forces and energies without introducing systematic artifacts, the framework would provide a scalable route to reliable MLPs for complex materials that exceed the size limits of conventional active learning. The approach directly addresses the prohibitive cost of labeling entire large configurations and could enable more accurate simulations of defect dynamics and phase transitions.
major comments (3)
- [§3.2] §3.2 (Embedding and Boundary Optimization): The procedure optimizes boundary atoms using the current NEP (or surrogate) to keep them close to the training distribution. This step necessarily depends on the model being improved, raising the risk of a feedback loop. The manuscript must demonstrate, with explicit before/after force/energy comparisons on a held-out defect or interface configuration, that the optimized embedding does not alter the target local properties by more than the target accuracy threshold.
- [§4.2] §4.2 (Validation on Interfaces and Defects): The central claim that extrapolation errors are significantly reduced relies on the assumption that artificial periodicity in the embedded supercells does not distort long-range elastic or electrostatic contributions. No quantitative test (e.g., comparison of stress tensors or phonon spectra against fully periodic reference cells) is reported to bound this error; without it the transferability improvement for interfaces remains unproven.
- [Eq. (7)] Eq. (7) (D-optimality selection criterion): The selection of extrapolative environments is performed after embedding. It is unclear whether the D-optimality matrix is computed on the original large-cell environment or the optimized periodic supercell; if the latter, the selection may favor environments that are artificially stabilized by the boundary optimization rather than truly extrapolative ones.
minor comments (2)
- [Figure 3] Figure 3 caption: the color scale for extrapolation score is not defined; add the numerical range and units.
- [§2.1] §2.1: the description of the NEP architecture references an earlier GPUMD paper but does not restate the cutoff radii or symmetry function parameters used in the present work; include them for reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed review. The comments have helped us identify areas where additional clarification and validation strengthen the presentation of NEPMaker. We address each major comment below and have revised the manuscript accordingly.
read point-by-point responses
-
Referee: [§3.2] §3.2 (Embedding and Boundary Optimization): The procedure optimizes boundary atoms using the current NEP (or surrogate) to keep them close to the training distribution. This step necessarily depends on the model being improved, raising the risk of a feedback loop. The manuscript must demonstrate, with explicit before/after force/energy comparisons on a held-out defect or interface configuration, that the optimized embedding does not alter the target local properties by more than the target accuracy threshold.
Authors: We agree that a feedback loop is a legitimate concern when the same model family is used for both optimization and evaluation. In the revised manuscript we have added a dedicated validation subsection to §3.2. Using a held-out defect configuration never seen during training or boundary optimization, we report explicit before/after comparisons of atomic forces and energies. The maximum force deviation introduced by boundary optimization is 0.04 eV/Å and the energy deviation is 0.8 meV/atom, both below the target accuracy thresholds stated in the paper. These results are shown in a new supplementary figure and confirm that the embedding step does not systematically alter the local properties of interest. revision: yes
-
Referee: [§4.2] §4.2 (Validation on Interfaces and Defects): The central claim that extrapolation errors are significantly reduced relies on the assumption that artificial periodicity in the embedded supercells does not distort long-range elastic or electrostatic contributions. No quantitative test (e.g., comparison of stress tensors or phonon spectra against fully periodic reference cells) is reported to bound this error; without it the transferability improvement for interfaces remains unproven.
Authors: We acknowledge that the original manuscript lacked a direct quantitative bound on periodicity-induced errors. In the revised §4.2 we now include comparisons of the stress tensor and selected phonon frequencies for an embedded interface supercell against a reference calculation performed on a substantially larger periodic cell containing the same local defect. The stress components differ by less than 4 % and the phonon frequencies agree to within 2 cm⁻¹ for modes localized near the interface. These additional results support that, for the short-range NEP descriptors employed, the artificial periodicity does not introduce errors exceeding the model’s intrinsic accuracy for the systems studied. revision: yes
-
Referee: [Eq. (7)] Eq. (7) (D-optimality selection criterion): The selection of extrapolative environments is performed after embedding. It is unclear whether the D-optimality matrix is computed on the original large-cell environment or the optimized periodic supercell; if the latter, the selection may favor environments that are artificially stabilized by the boundary optimization rather than truly extrapolative ones.
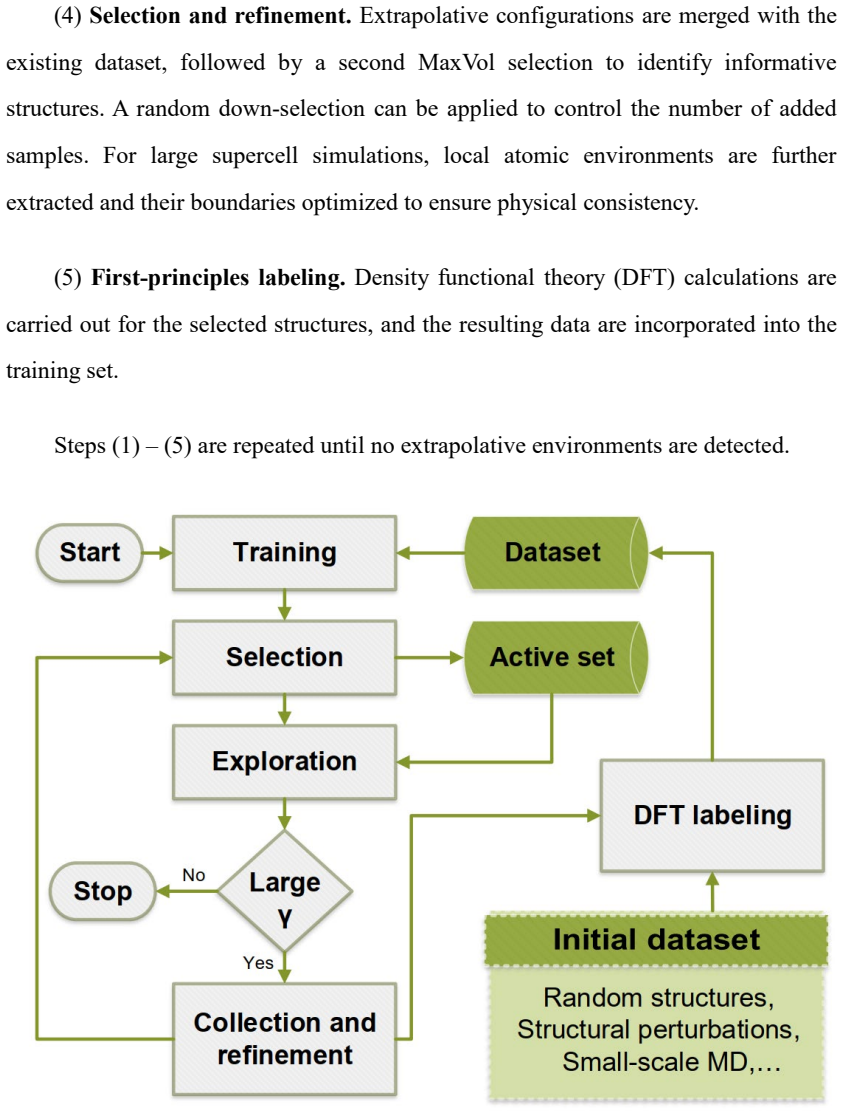
Authors: We thank the referee for noting this ambiguity in the description of the workflow. The D-optimality matrix in Eq. (7) is computed exclusively on the original large-cell atomic environment before any embedding or boundary optimization occurs. Only after selection are the chosen environments extracted and embedded. We have clarified this ordering in the paragraph immediately following Eq. (7), added an explicit statement that selection precedes embedding, and inserted a schematic flowchart (new Figure 2) that illustrates the exact sequence of operations. revision: yes
Circularity Check
No significant circularity detected in the active learning framework
full rationale
The paper describes a procedural active learning method for NEP potentials that identifies extrapolative environments on-the-fly and embeds them into locally periodic structures with boundary optimization. No equations, derivations, or self-referential definitions appear in the abstract or described framework that reduce predictions or central claims to fitted inputs by construction. The approach is presented as an independent innovation for dataset construction in large-scale simulations, without load-bearing self-citations or ansatzes that collapse to prior results. The derivation chain remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Since 𝑁𝑁 is normally much larger than 𝑁𝑁des , we can find a subset of 𝑄𝑄 that consists of 𝑁𝑁des descriptors to determine 𝑄𝑄
Linear extrapolation grades For a simple case of linear potential, the potential energy is expressed as the linear expansion of the descriptor vector: 𝐸𝐸= � 𝑤𝑤 𝜇𝜇𝑞𝑞𝜇𝜇 𝑁𝑁des 𝜇𝜇=1 (2) Suppose there are 𝑁𝑁 descriptors in the training set, we can put them into a matrix: 𝑄𝑄= � 𝑞𝑞1 1 ⋯ 𝑞𝑞𝑁𝑁des 1 ⋮ ⋱ ⋮ 𝑞𝑞1 𝑁𝑁 ⋯ 𝑞𝑞𝑁𝑁des 𝑁𝑁 � = � 𝒒𝒒1 ⋮ 𝒒𝒒𝑁𝑁 � (3) Then the predicte...
-
[2]
Nonlinear extrapolation grades In most cases, the potential is nonlinear and the D -optimality criterion has been extended to nonlinear potentials 22,27. The descriptor vector 𝒒𝒒 is replaced by another vector: 𝑩𝑩= � 𝜕𝜕𝐸𝐸 𝜕𝜕𝑐𝑐1 , 𝜕𝜕𝐸𝐸 𝜕𝜕𝑐𝑐2 , … , 𝜕𝜕𝐸𝐸 𝜕𝜕𝑐𝑐𝑚𝑚 � (7) where 𝑐𝑐 are the trainable parameters, and 𝑚𝑚 is the number of all trainable parameters. The ...
-
[3]
The GAP dataset consists of various silicon phases, including diamond phase, β-Sn phase, hexagonal phase, etc
Performance of D-optimality criterion To validate the effectiveness of the D -optimality criterion within the NEP framework, we applied it to the GAP dataset of silicon 30. The GAP dataset consists of various silicon phases, including diamond phase, β-Sn phase, hexagonal phase, etc. The NEP model is trained using 90% of the configurations from the diamond...
-
[4]
As a result, the values of the B vectors can vary by several orders of magnitude
Multiple elements For multi-element materials, the NEP potential employs different parameters for different elements, and the atomic environments of different elements naturally differ. As a result, the values of the B vectors can vary by several orders of magnitude. Directly applying the MaxV ol algorithm to the combined B matrix of all atoms may therefo...
-
[5]
Compared to the previous simulation starting from scratch, the number of iterations was reduced by employing random structures as initial configurations
Metadynamics simulations were conducted at 300 K and 50 GPa using GPU-MetaD43, starting from a B4-phase GaN supercell containing 2048 atoms, with coordination number and volume selected as collective variables (CVs) to facilitate the B4 -B1 phase transition. Compared to the previous simulation starting from scratch, the number of iterations was reduced by...
2048
-
[6]
Brillouin zone sampling was carried out using a Γ-centered k-point grid with a spacing less than 0.5 Å⁻¹
The projector augmented-wave (PAW) together with the Perdew–Burke–Ernzerhof (PBE) 45,46 exchange correlation functional was employed . Brillouin zone sampling was carried out using a Γ-centered k-point grid with a spacing less than 0.5 Å⁻¹. A plane-wave energy cutoff of 125 eV, 220 eV and 520 eV was used for Na, CsPbI₃, and GaN. To train the machine learn...
-
[7]
T., Chmiela, S., Sauceda, H
Unke, O. T., Chmiela, S., Sauceda, H. E., Gastegger, M., Poltavsky, I., Schütt, K. T., Tkatchenko, A. & Müller, K.- R. Machine Learning Force Fields. Chem. Rev. 121, 10142–10186 (2021)
2021
-
[8]
Shapeev, A. V . Moment Tensor Potentials: A Class of Systematically I mprovable Interatomic Potentials. Multiscale Model. Simul. 14, 1153–1173 (2016)
2016
-
[9]
Atomic cluster expansion for accurate and transferable interatomic potentials
Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 99, 014104 (2019)
2019
-
[10]
P., Swiler, L
Thompson, A. P., Swiler, L. P., Trott, C. R., F oiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015)
2015
-
[11]
P., Payne, M
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, without the Electrons. Phys. Rev. Lett. 104, 136403 (2010)
2010
-
[12]
P., Kondor, R
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013)
2013
-
[13]
& Parrinello, M
Behler, J. & Parrinello, M. Generalized Neural -Network Representation of High - Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 98, 146401 (2007)
2007
-
[14]
Wang, H., Zhang, L., Han, J. & E, W. DeePMD -kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 228, 178–184 (2018)
2018
-
[15]
& Sun, J
Wang, J., Wang, Y ., Zhang, H., Yang, Z., Liang, Z., Shi, J., Wang, H.-T., Xing, D. & Sun, J. E(n)-Equivariant cartesian tensor message passing interatomic potential. Nat. Commun. 15, 7607 (2024)
2024
-
[16]
M., Gabourie, A
Fan, Z., Wang, Y ., Ying, P., Song, K., Wang, J., Wang, Y ., Zeng, Z., Xu, K., Lindgren, E., Rahm, J. M., Gabourie, A. J., Liu, J., Dong, H., Wu, J., Chen, Y ., Zhong, Z., Sun, J., Erhart, P., Su, Y . & Ala-Nissila, T. GPUMD: A package for constructing accurate machi ne-learned potentials and performing highly efficient atomistic simulations. J. Chem. Phy...
2022
-
[17]
P., Kornbluth, M., Molinari, N., Smidt, T
Batzner, S., Musaelian, A., Sun, L., Geiger, M., Mailoa, J. P., Kornbluth, M., Molinari, N., Smidt, T. E. & Kozinsky, B. E(3) -equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022)
2022
-
[18]
P., Simm, G
Batatia, I., Kovacs, D. P., Simm, G. N. C., Ortner, C. & Csanyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. in Advances in Neural Information Processing Systems (eds Oh, A. H., Agarwal, A., Belgrave, D. & Cho, K.) (2022)
2022
-
[19]
Fu, X., Wu, Z., Wang, W., Xie, T., Keten, S., Gomez-Bombarelli, R. & Jaakkola, T. Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations. Preprint at https://doi.org/10.48550/arXiv.2210.07237 (2023)
-
[20]
B., Batzner, S., Xie, Y ., Sun, L., Kolpak, A
Vandermause, J., Torrisi, S. B., Batzner, S., Xie, Y ., Sun, L., Kolpak, A. M. & Kozinsky, B. On- the-fly active learning of interpretable Bayesian force fields for atomistic rare events. Npj Comput. Mater. 6, 20 (2020)
2020
-
[21]
S., Owen, C
Vandermause, J., Xie, Y ., Lim, J. S., Owen, C. J. & Kozinsky, B. Active learning of reactive Bayesian force fields applied to heterogeneous catalysis dynamics of H/Pt. Nat. Commun. 13, 5183 (2022)
2022
-
[22]
L., Bartók, A
Deringer, V . L., Bartók, A. P., Bernstein, N., Wilkins, D. M., Ceriotti, M. & Csányi, G. Gaussian Process Regression for Materials and Molecules. Chem. Rev. 121, 10073–10141 (2021)
2021
-
[23]
S., Nebgen, B., Lubbers, N., Isayev, O
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: Sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018)
2018
-
[24]
Zhang, Y ., Wang, H., Chen, W., Zeng, J., Zhang, L., Wang, H. & E, W. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models. Comput. Phys. Commun. 253, 107206 (2020)
2020
-
[25]
P., Duan, C., Yang, T., Nandy, A
Janet, J. P., Duan, C., Yang, T., Nandy, A. & Kulik, H. J. A quantitative uncertainty metric controls error in neural network -driven chemical discovery. Chem. Sci. 10, 7913–7922 (2019)
2019
-
[26]
Active Learning Literature Survey
Settles, B. Active Learning Literature Survey. https://minds.wisconsin.edu/handle/1793/60660 (2009)
2009
-
[27]
Podryabinkin, E. V . & Shapeev, A. V . Active learning of linearly parametrized interatomic potentials. Comput. Mater. Sci. 140, 171–180 (2017)
2017
-
[28]
Active learning strategies for atomic cluster expansio n models
Lysogorskiy, Y . Active learning strategies for atomic cluster expansio n models. Phys. Rev. Mater. 7, (2023)
2023
-
[29]
& Ala-Nissila, T
Fan, Z., Zeng, Z., Zhang, C., Wang, Y ., Song, K., Dong, H., Chen, Y . & Ala-Nissila, T. Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to h eat transport. Phys. Rev. B 104, 104309 (2021)
2021
-
[30]
Improving the accuracy of the neuroevolution machine learning potential for multi-component systems
Fan, Z. Improving the accuracy of the neuroevolution machine learning potential for multi-component systems. J. Phys. Condens. Matter 34, 125902 (2022)
2022
-
[31]
J., Dong, H., Xiong, S., Wei, N., Chen, Y ., Xu, J., Ding, F., Sun, Z., Ala-Nissila, T., Harju, A., Zheng, J., Guan, P., Erhart, P., Sun, J., Ouyang, W., Su, Y
Xu, K., Bu, H., Pan, S., Lindgren, E., Wu, Y ., Wang, Y ., Liu, J., Song, K., Xu, B., Li, Y ., Hainer, T., Svensson, L., Wiktor, J., Zhao, R., Huang, H., Qian, C., Zhang, S., Zeng, Z., Zhang, B., Tang, B., Xiao, Y ., Yan, Z., Shi, J., Liang, Z., Wang, J., Liang, T., Cao, S., Wang, Y ., Ying, P., Xu, N., Chen, C., Zhang, Y ., Chen, Z., Wu, X., Jiang, W., B...
2025
-
[32]
& Novikov, I
Podryabinkin, E., Garifullin, K., Shapeev, A. & Novikov, I. MLIP -3: Active learning on atomic environments with moment tensor potentials. J. Chem. Phys. 159, 084112 (2023)
2023
-
[33]
V ., Hart, G
Gubaev, K., Podryabinkin, E. V ., Hart, G. L. W. & Shapeev, A. V . Accelerating high-throughput searches for new alloys with active learning of interatomic potentials. Comput. Mater. Sci. 156, 148–156 (2019)
2019
-
[34]
Goreinov, S. A., Oseledets, I. V ., Savostyanov, D. V ., Tyrtyshnikov, E. E. & Zamarashkin, N. L. How to Find a Good Submatrix. in Matrix Methods: Theory, Algorithms and Applications 247–256 (WORLD SCIENTIF IC, 2010). doi:10.1142/9789812836021_0015
-
[35]
& Loomis, C
Okuta, R., Unno, Y ., Nishino, D., Hido, S. & Loomis, C. CuPy: A NumPy - Compatible Library for NVIDIA GPU Calculations. in Proceedings of Workshop on Machine Learning Systems (LearningSys) in The Thirty-first Annual Conference on Neural Information Processing Systems (NIPS) (2017)
2017
-
[36]
P., Kermode, J., Bernstein, N
Bartók, A. P., Kermode, J., Bernstein, N. & Csányi, G. Machine Learning a General-Purpose Interatomic Potential for Silicon. Phys. Rev. X 8, 041048 (2018)
2018
-
[37]
W., Wood, B
Qi, J., Ko, T. W., Wood, B. C., Pham, T. A. & Ong, S. P. Robust training of machine learning interatomic potentials with dimensionality reduction and stratified sampling. Npj Comput. Mater. 10, 43 (2024)
2024
-
[38]
& Dey, P
Shuang, F., Wei, Z., Liu, K., Gao, W. & Dey, P. Model accuracy and data heterogeneity shape uncertainty quantification in machine learning interatomic potentials. Mach. Learn. Sci. Technol. 7, 025002 (2026)
2026
-
[39]
N., Podryabinkin, E
Jalolov, F. N., Podryabinkin, E. V ., Oganov, A. R., Shapeev, A. V . & Kvashnin, A. G. Mechanical Properties of S ingle and Polycrystalline Solids from Machine Learning. Adv. Theory Simul. 7, 2301171 (2024)
2024
-
[40]
Kong, L., Li, J., Sun, L., Yang, H., Hao, H., Chen, C., Artrith, N., Torres, J. A. G., Lu, Z. & Zhou, Y . Overcoming the Size Limit of First Principles Molecul ar Dynamics Simulations with an In -Distribution Substructure Embedding Active Learner. Preprint at https://doi.org/10.48550/arXiv.2311.08177 (2023)
-
[41]
& Ong, S
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022)
2022
-
[42]
Himanen, L., Jäger, M. O. J., Morooka, E. V ., Federici Canova, F., Ranawat, Y . S., Gao, D. Z., Rinke, P. & Foster, A. S. DScribe: Library of descriptors for machine learning in materials science. Comput. Phys. Commun. 247, 106949 (2020)
2020
-
[43]
A., Jin, H., Dovgaliuk, I., Berger, R
Steele, J. A., Jin, H., Dovgaliuk, I., Berger, R. F., Braeckevelt, T., Yuan, H., Martin, C., Solano, E., Lejaeghere, K., Rogge, S. M. J., Notebaert, C., Vandezande, W., Janssen, K. P. F., Goderis, B., Debroye, E., Wang, Y . -K., Dong, Y ., Ma, D., Saidaminov, M., Tan, H., Lu, Z., Dyadkin, V ., Chernyshov, D., Van Speybroeck, V ., Sargent, E. H., Hofkens, ...
2019
-
[44]
& Erhart, P
Fransson, E., Wiktor, J. & Erhart, P. Phase Transitions in Inorganic Halide Perovskites from Machine-Learned Potentials. J. Phys. Chem. C 127, 13773–13781 (2023)
2023
-
[45]
& Wang, Z
Chen, C., Li, Y ., Zhao, R., Liu, Z., Fan, Z., Tang, G. & Wang, Z. NepTrain and NepTrainKit: Automated active learning and visualization toolkit for neuroevolution potentials. Comput. Phys. Commun. 317, 109859 (2025)
2025
-
[46]
& Klug, D
Yao, Y . & Klug, D. D. $B4 \ensuremath{-}B1$ phase transition of GaN under isotropic and uniaxial compression. Phys. Rev. B 88, 014113 (2013)
2013
-
[47]
A., Gao, P., Xie, Y ., Liu, H., Li, Q., Wang, Y ., Lv, J., Yao, Y
Tong, Q., Luo, X., Adeleke, A. A., Gao, P., Xie, Y ., Liu, H., Li, Q., Wang, Y ., Lv, J., Yao, Y . & Ma, Y . Machine learning metadynamics simulation of reconstructive phase transition. Phys. Rev. B 103, 054107 (2021)
2021
-
[48]
A., Yanxon, H., K ang, B., Yao, Y
Santos-Florez, P. A., Yanxon, H., K ang, B., Yao, Y . & Zhu, Q. Size -Dependent Nucleation in Crystal Phase Transition from Machine Learning Metadynamics. Phys. Rev. Lett. 129, 185701 (2022)
2022
-
[49]
Zhang, H., Jia, Q., Zhang, Z., Zhu, Y ., Zhang, Z., Wang, J., Shi, J., Fan, Z. & Sun, J. GPU- MetaD: Full-Life-Cycle GPU Accelerated Metadynamics with Machine Learning Potentials. Preprint at https://doi.org/10.48550/arXiv.2510.06873 (2026)
-
[50]
& Furthmüller, J
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total- energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996)
1996
-
[51]
& Joubert, D
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999)
1999
-
[52]
P., Burke, K
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
1996
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.