Recognition: unknown
Accurate and Efficient Interatomic Potentials for Dislocations in InP
Pith reviewed 2026-05-10 00:17 UTC · model grok-4.3
The pith
Bespoke machine-learned potentials reproduce partial dislocation formation energies in InP to within 4% of DFT.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
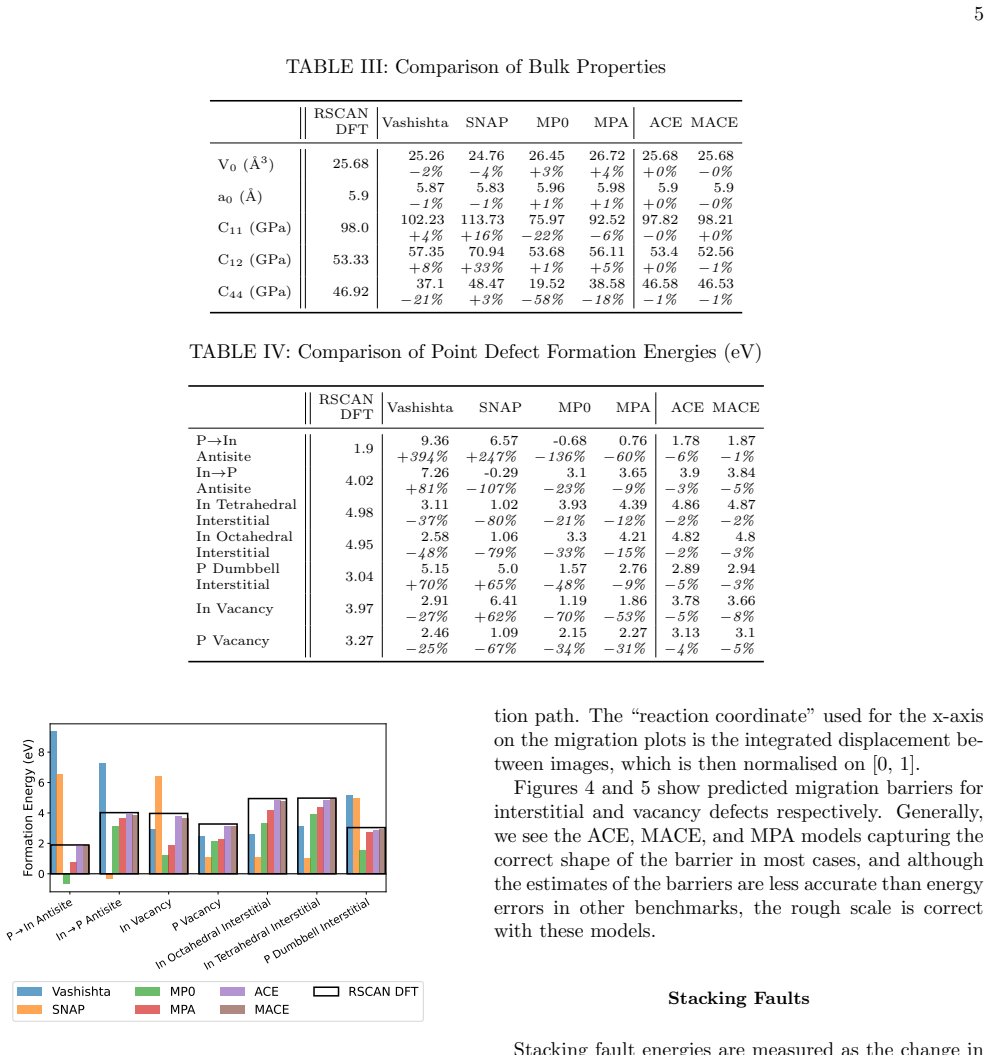
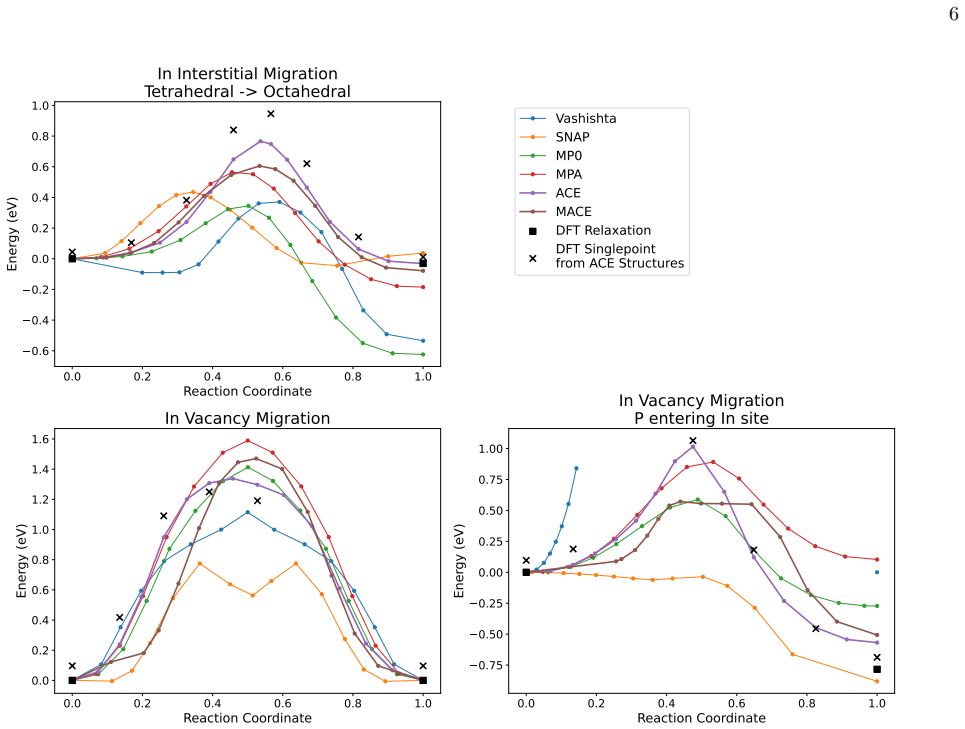
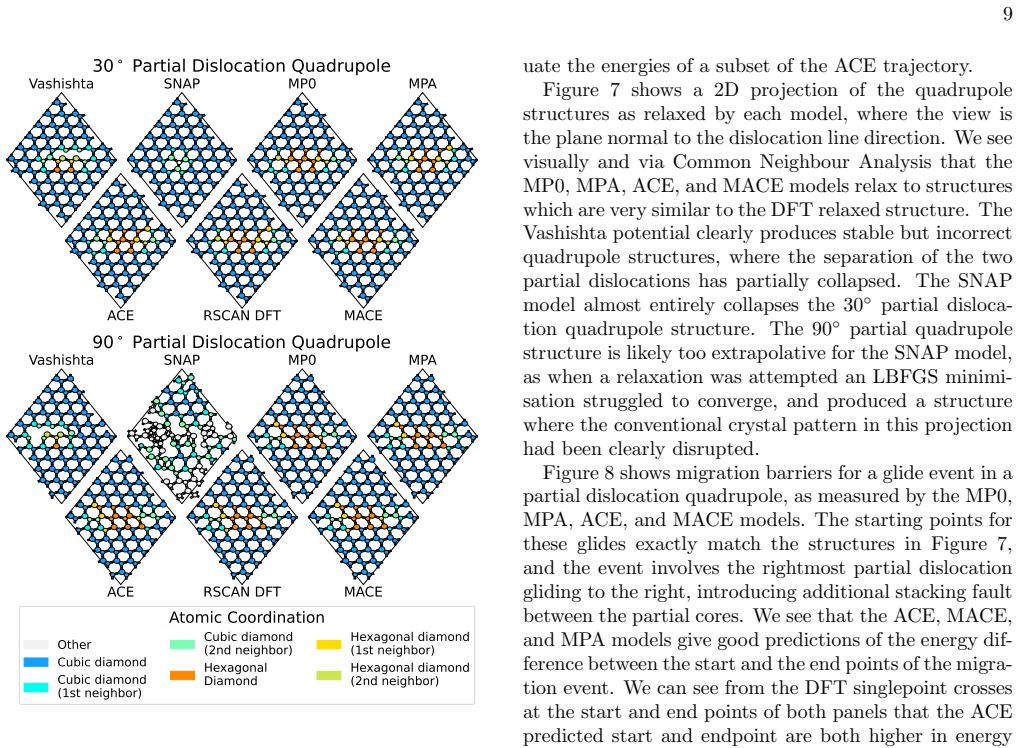
A DFT dataset focused on dislocation-relevant atomic arrangements in InP is used to train ACE and MACE models that achieve at most 4% error on partial dislocation formation energies, compared with 18% for the MACE-MPA foundation model and 42-50% for previously published potentials; the custom MACE model delivers this accuracy at roughly five times the evaluation speed of the MP0 and MPA models.
What carries the argument
Atomic Cluster Expansion (ACE) and MACE machine-learned interatomic potentials trained on a dislocation-specific DFT dataset and validated against RSCAN calculations.
If this is right
- Simulations of dislocation mobility and interactions in InP can now be performed with formation-energy errors below 4% relative to DFT.
- The fivefold speed-up of the bespoke MACE model enables larger-scale or longer-time simulations than are practical with the slower foundation models.
- Direct comparison to literature potentials shows that domain-specific training data reduces errors on defect energetics by more than an order of magnitude in this material.
- The same training and validation workflow can be reused to produce potentials for other defect-driven properties once additional DFT data are added.
Where Pith is reading between the lines
- The dataset-construction strategy used here could be applied to other compound semiconductors to obtain similarly accurate potentials for their dislocations.
- Faster potentials open the possibility of coupling atomistic dislocation simulations to continuum models of plastic flow in device-scale structures.
- If the models also reproduce experimental dislocation velocities, they could reduce reliance on direct DFT for screening doping or alloying effects on InP mechanical behavior.
Load-bearing premise
The new DFT dataset and validation tests include enough of the atomic environments that actually occur during dislocation motion and core reconstruction in InP for the reported energy errors to carry over to dynamic simulations.
What would settle it
If molecular-dynamics runs with these potentials produce dislocation core structures, migration barriers, or velocities that differ substantially from independent RSCAN DFT calculations or from experimental measurements on InP, the accuracy claim would be refuted.
Figures





read the original abstract
We present Atomic Cluster Expansion (ACE) and MACE models trained on a new dataset of Density Functional Theory (DFT) calculations, constructed for the task of studying the mobility of dislocations in Indium Phosphide (InP). The models are validated in a suite of tests against RSCAN DFT, and compared with previously published potentials from literature. Our new models act as much better surrogates for DFT than the literature models: errors on partial dislocation formation energies are at most 4% for both ACE and MACE, compared with 18% for the MACE-MPA foundation model and 42-50% for earlier bespoke potentials. The bespoke MACE model achieves this accuracy while being around five times faster to evaluate than the MP0 and MPA foundation models.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents ACE and MACE interatomic potentials trained on a new DFT dataset for InP, constructed specifically to study dislocation mobility. The models are validated against RSCAN DFT and compared to literature potentials, with the central claim that both achieve at most 4% error on partial-dislocation formation energies (versus 18% for MACE-MPA and 42-50% for earlier bespoke models) while the bespoke MACE model evaluates approximately five times faster than MP0/MPA foundation models.
Significance. If the reported accuracy on formation energies generalizes, the potentials would offer a clear improvement as efficient DFT surrogates for large-scale dislocation simulations in InP, a key semiconductor material. The speed advantage over foundation models is a notable practical strength for molecular-dynamics applications.
major comments (2)
- [Abstract] Abstract: the claim that the models are 'constructed for the task of studying the mobility of dislocations' rests on validation limited to static partial-dislocation formation energies. No tests on Peierls barriers, nudged-elastic-band paths, or shear-induced saddle-point configurations are described, which is load-bearing for transferability to mobility and core dynamics.
- [Validation suite] Validation suite (results section): the reported error metrics (at most 4%) are given only for equilibrium formation energies; without explicit coverage of high-strain or thermally activated configurations in the training or test sets, the generalization to dislocation glide cannot be assessed from the presented evidence.
minor comments (2)
- [Abstract] The abstract and introduction could more explicitly qualify the scope of the validation (static vs. dynamic properties) to avoid overstatement of applicability to mobility studies.
- [Results] Table or figure presenting the formation-energy comparisons should include the number of configurations tested and the precise definition of the error metric (e.g., relative to what reference value) for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments on our manuscript. We have addressed each major point below, clarifying the scope of our validation while acknowledging its limitations. Revisions have been made to the abstract and results section to better align the claims with the presented evidence.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that the models are 'constructed for the task of studying the mobility of dislocations' rests on validation limited to static partial-dislocation formation energies. No tests on Peierls barriers, nudged-elastic-band paths, or shear-induced saddle-point configurations are described, which is load-bearing for transferability to mobility and core dynamics.
Authors: We appreciate the referee highlighting this distinction. The dataset was constructed with a focus on dislocation core configurations in InP, and the abstract phrasing reflected the broader motivation for developing accurate potentials suitable for large-scale dislocation studies. However, we agree that the validation is restricted to static partial-dislocation formation energies and does not include tests on Peierls barriers, NEB paths, or saddle-point configurations. To address this, we have revised the abstract to state that the models are developed and validated for accurate modeling of dislocation core structures and formation energies, serving as a foundation for subsequent mobility investigations. A new paragraph has been added to the discussion section explicitly noting the absence of dynamic barrier calculations and the need for such tests to confirm transferability to glide processes. revision: partial
-
Referee: [Validation suite] Validation suite (results section): the reported error metrics (at most 4%) are given only for equilibrium formation energies; without explicit coverage of high-strain or thermally activated configurations in the training or test sets, the generalization to dislocation glide cannot be assessed from the presented evidence.
Authors: The primary error metrics focus on equilibrium formation energies because these directly assess the potentials' fidelity in reproducing the energetics and structures of partial dislocation cores, which is a critical prerequisite for any dislocation-related simulation. The training set incorporates configurations sampled from dislocation models, which include a range of local atomic environments and moderate strains around the cores. We nevertheless agree that the test suite does not explicitly cover high-strain regimes or thermally activated saddle points, limiting direct claims about generalization to dislocation glide. We have expanded the results section to provide more detail on the strain distributions present in the training data and have inserted a limitations paragraph stating that additional validation on glide barriers would be required to fully assess performance in dynamic mobility contexts. revision: partial
Circularity Check
No circularity in derivation or validation chain.
full rationale
The paper constructs a new DFT dataset, trains ACE and MACE interatomic potentials on it, and reports direct numerical errors on held-out partial-dislocation formation energies against independent RSCAN DFT reference calculations. No equations, fitted parameters, or self-citations reduce the reported formation-energy errors to the training inputs by construction; the comparisons are standard out-of-sample validation against external DFT data. The central claim of improved surrogate accuracy therefore rests on empirical test-set performance rather than tautological re-expression of the training data.
Axiom & Free-Parameter Ledger
free parameters (1)
- ACE and MACE model hyperparameters and weights
axioms (1)
- domain assumption DFT calculations (RSCAN functional) provide sufficiently accurate reference data for training and validation of interatomic potentials.
Reference graph
Works this paper leans on
-
[1]
64x0e + 64x1o
was used to generate the stacking fault and disloca- tion quadrupole structures. Table I provides an overview of the contents of the dataset, partitioned by Configuration Type, which pro- vides a concise label for the property or application a particular structure was designed to target. The table also includes information about the number of structures, ...
1975
-
[2]
Displacment (Å) 0.0 0.5 1.0 1.5 2.0Energy Density (J/m2) (001)[100] Stacking Fault 0 1 2 3 4
-
[3]
Displacment (Å) 0.00 0.25 0.50 0.75 1.00 1.25 1.50 1.75 2.00Energy Density (J/m2) (001)[110] Stacking Fault 0 1 2 3 4
-
[4]
Displacment (Å) 0.0 0.2 0.4 0.6 0.8 1.0 1.2Energy Density (J/m2) (110)[110] Stacking Fault 0.0 0.5 1.0 1.5 2.0 2.5
-
[5]
6: Comparison of predicted stacking fault barriers for several InP potentials
Displacment (Å) 0.0 0.2 0.4 0.6 0.8 1.0 1.2Energy Density (J/m2) (111)[112] Stacking Fault FIG. 6: Comparison of predicted stacking fault barriers for several InP potentials. The four larger panels show the predicted stacking fault energy curve as a function of the displacement of the upper surface, for four different stacking faults. The twelve smaller p...
-
[6]
Tomiya, T
S. Tomiya, T. Hino, S. Goto, M. Takeya, and M. Ikeda, Dislocation related issues in the degradation of gan-based laser diodes, IEEE Journal of Selected Topics in Quan- tum Electronics10, 1277 (2004)
2004
-
[7]
O’Hara, P
S. O’Hara, P. W. Hutchinson, and P. S. Dob- son, The origin of dislocation climb during laser operation, Applied Physics Letters30, 368 (1977), https://pubs.aip.org/aip/apl/article- pdf/30/8/368/18434635/368 1 online.pdf
1977
-
[8]
S. L. Frederiksen and K. W. Jacobsen, Density func- tional theory studies of screw dislocation core structures in bcc metals, Philosophical Magazine83, 365 (2003), https://doi.org/10.1080/0141861021000034568
-
[9]
Ventelon and F
L. Ventelon and F. Willaime, Core structure and peierls potential of screw dislocations inα-fe from first prin- ciples: cluster versus dipole approaches, Journal of Computer-Aided Materials Design14, 85 (2007)
2007
-
[10]
Clouet, L
E. Clouet, L. Ventelon, and F. Willaime, Dislocation core energies and core fields from first principles, Phys. Rev. Lett.102, 055502 (2009)
2009
-
[11]
S. P. Beckman, X. Xu, P. Specht, E. R. Weber, C. Kisielowski, and D. C. Chrzan, Ab initio prediction of the structure of glide set dislocation cores in gaas, Journal of Physics: Condensed Matter14, 12673 (2002)
2002
-
[12]
S. Rao, C. Hernandez, J. P. Simmons, T. A. Parthasarathy, and C. Woodward, Green’s func- tion boundary conditions in two-dimensional and three-dimensional atomistic simulations of disloca- tions, Philosophical Magazine A77, 231 (1998), https://doi.org/10.1080/01418619808214240
-
[13]
D. R. Trinkle, Lattice green function for extended de- fect calculations: Computation and error estimation with long-range forces, Phys. Rev. B78, 014110 (2008)
2008
-
[14]
Y. Liu, G. Lu, Z. Chen, and N. Kioussis, An improved qm/mm approach for metals, Modelling and Simulation in Materials Science and Engineering15, 275 (2007)
2007
-
[15]
Zhao and G
Y. Zhao and G. Lu, Qm/mm study of disloca- tion—hydrogen/helium interactions inα-fe, Modelling and Simulation in Materials Science and Engineering19, 065004 (2011)
2011
-
[16]
Grigorev, T
P. Grigorev, T. D. Swinburne, and J. R. Kermode, Hybrid quantum/classical study of hydrogen-decorated screw dislocations in tungsten: Ultrafast pipe diffusion, core reconstruction, and effects on glide mechanism, Phys. Rev. Mater.4, 023601 (2020)
2020
-
[17]
P. S. Branicio, J. P. Rino, C. K. Gan, and H. Tsuzuki, In- teraction potential for indium phosphide: a molecular dy- namics and first-principles study of the elastic constants, generalized stacking fault and surface energies, Journal of Physics: Condensed Matter21, 095002 (2009)
2009
-
[18]
M. A. Cusentino, M. A. Wood, and A. P. Thompson, Explicit multielement extension of the spectral neighbor analysis potential for chemically complex systems, The Journal of Physical Chemistry A124, 5456 (2020)
2020
-
[19]
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, S. M. Blau, V. C˘ arare, J. P. Darby, S. De, F. D. Pia, V. L. Deringer, R. Elijoˇ sius, Z. El-Machachi, F. Falcioni, E. Fako, A. C. Ferrari, A. Genreith-Schriever, 12 J. George, R. E. A. Goo...
-
[20]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, and K. A. Persson, Commentary: The materials project: A materials genome approach to accelerating materials innovation, APL Materials1, 011002 (2013), https://pubs.aip.org/aip/apm/article- pdf/doi/10.1063/1.4812323/13163869/011002 1 online.pdf
-
[21]
Barroso-Luque et al., Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
L. Barroso-Luque, M. Shuaibi, X. Fu, B. M. Wood, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick, and Z. W. Ulissi, Open materials 2024 (omat24) inorganic materi- als dataset and models (2024), arXiv:2410.12771 [cond- mat.mtrl-sci]
-
[22]
Schmidt, T
J. Schmidt, T. F. Cerqueira, A. H. Romero, A. Loew, F. J¨ ager, H.-C. Wang, S. Botti, and M. A. Marques, Improving machine-learning models in materials science through large datasets, Materials Today Physics48, 101560 (2024)
2024
-
[23]
Celli, M
V. Celli, M. Kabler, T. Ninomiya, and R. Thomson, The- ory of dislocation mobility in semiconductors, Phys. Rev. 131, 58 (1963)
1963
-
[24]
Bullough and R
R. Bullough and R. C. Newman, The kinetics of migra- tion of point defects to dislocations, Reports on Progress in Physics33, 101 (1970)
1970
-
[25]
R. Vardya and S. Mahajan, Mechanism of dislo- cation climb in binary and mixed iii-v semicon- ductors, Philosophical Magazine A71, 465 (1995), https://doi.org/10.1080/01418619508244462
-
[26]
A. T. Blumenau, M. I. Heggie, C. J. Fall, R. Jones, and T. Frauenheim, Dislocations in diamond: Core structures and energies, Phys. Rev. B65, 205205 (2002)
2002
-
[27]
De Cooman, C
B. De Cooman, C. Carter, C. Kam Toi, and J. Shealy, The characterization of misfit dislocations at 100 hetero- junctions in iii–v compound semiconductors, Acta Met- allurgica37, 2779 (1989)
1989
-
[28]
Allen and A
C. Allen and A. P. Bart´ ok, Optimal data generation for machine learned interatomic potentials, Machine Learn- ing: Science and Technology3, 045031 (2022)
2022
-
[29]
J. H. Lloyd-Williams and B. Monserrat, Lattice dynam- ics and electron-phonon coupling calculations using non- diagonal supercells, Phys. Rev. B92, 184301 (2015)
2015
-
[30]
Grigorev, L
P. Grigorev, L. Fr´ erot, F. Birks, A. Gola, J. Golebiowski, J. Grießer, J. L. H¨ ormann, A. Klemenz, G. Moras, W. G. N¨ ohring, J. A. Oldenstaedt, P. Patel, T. Reichenbach, T. Rocke, L. Shenoy, M. Walter, S. Wengert, L. Zhang, J. R. Kermode, and L. Pastewka, matscipy: materials science at the atomic scale with python, Journal of Open Source Software9, 56...
2024
-
[31]
S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson, and M. Payne, First principles methods using CASTEP, Z. Kristall.220, 567 (2005)
2005
-
[32]
A. P. Bart´ ok and J. R. Yates, Regularized SCAN functional, The Journal of Chemical Physics150, 161101 (2019), https://pubs.aip.org/aip/jcp/article- pdf/doi/10.1063/1.5094646/13514553/161101 1 online.pdf
-
[33]
Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys
D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B41, 7892 (1990)
1990
-
[34]
Hasnip and C
P. Hasnip and C. Pickard, Electronic energy minimisa- tion with ultrasoft pseudopotentials, Computer Physics Communications174, 24 (2006)
2006
-
[35]
H. J. Monkhorst and J. D. Pack, Special points for brillouin-zone integrations, Phys. Rev. B13, 5188 (1976)
1976
-
[36]
Hjorth Larsen, J
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Du lak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasb- jerg, S. Lysgaard, J. Bergmann Maronsson, T. Max- son, T. Olsen, L. Pastewka, A. Peterson, C. Ros...
2017
-
[37]
Nichols, D
D. Nichols, D. Rimai, and R. Sladek, Elastic anharmonic- ity of inp: Its relationship to the high pressure transition, Solid State Communications36, 667 (1980)
1980
-
[38]
V. P. Vasil’ev and J.-C. Gachon, Thermodynamic prop- erties of inp, Inorganic Materials42, 1171 (2006)
2006
-
[39]
F. S. Hickernell and W. R. Gayton, Elastic constants of single-crystal indium phosphide, Journal of Applied Physics37, 462 (1966)
1966
-
[40]
F. A. Cunnel and E. W. Saker, Properties of iii-v com- pound semiconductors, Progress in Solids2, 37 (1957)
1957
-
[41]
Vurgaftman, J
I. Vurgaftman, J. Meyer, and R. Ram-Mohan, Band pa- rameters for iii-v compound semiconductors and their al- loys, J. Appl. Phys.89, 5815 (2001)
2001
-
[42]
Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys
R. Drautz, Atomic cluster expansion for accurate and transferable interatomic potentials, Phys. Rev. B99, 014104 (2019)
2019
-
[43]
Batatia, D
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Csanyi, Mace: Higher order equivariant message pass- ing neural networks for fast and accurate force fields, inAdvances in Neural Information Processing Systems, Vol. 35, edited by S. Koyejo, S. Mohamed, A. Agarwal, D. Belgrave, K. Cho, and A. Oh (Curran Associates, Inc.,
-
[44]
W. C. Witt, C. van der Oord, E. Gelˇ zinyt˙ e, T. J¨ arvinen, A. Ross, J. P. Darby, C. H. Ho, W. J. Baldwin, M. Sachs, J. Kermode, N. Bernstein, G. Cs´ anyi, and C. Ortner, Acepotentials.jl: A julia implementation of the atomic cluster expansion, The Journal of Chemical Physics159, 164101 (2023)
2023
-
[45]
Izmailov, D
P. Izmailov, D. Podoprikhin, T. Garipov, D. P. Vetrov, and A. G. Wilson, Averaging weights leads to wider optima and better generalization, inProceedings of the Thirty-Fourth Conference on Uncertainty in Artificial In- telligence, UAI 2018, Monterey, California, USA, August 6-10, 2018, edited by A. Globerson and R. Silva (AUAI Press, 2018) pp. 876–885
2018
-
[46]
P. H. Borcherds, G. F. Alfrey, A. D. B. Woods, and D. H. Saunderson, Phonon dispersion curves in indium phos- phide, Journal of Physics C: Solid State Physics8, 2022 13 (1975)
2022
-
[47]
Freysoldt, B
C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti, and C. G. Van de Walle, First- principles calculations for point defects in solids, Rev. Mod. Phys.86, 253 (2014)
2014
-
[48]
A. Stukowski, Visualization and analysis of atomistic simulation data with ovito–the open visualization tool, Modelling and Simulation in Materials Science and En- gineering18, 015012 (2009)
2009
-
[49]
T. Rocke and J. Kermode, Inp mlip testing framework, 10.5281/zenodo.16904066 (2025)
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.