Charge order, domain order, ideal mixing and absence of demixing in 2D binary mixtures of alcohols
Pith reviewed 2026-05-08 09:08 UTC · model grok-4.3
The pith
Two-dimensional alcohol mixtures mix ideally without the phase separation seen in three dimensions, due to charge ordering.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Binary mixtures of two dimensional site-based models of alcohols exhibit ideal mixing and a competition between ideality and micro phase separation in chain-like polar head aggregates, which cannot be explained by enhanced fluctuations alone but point to charge ordering shaping the local structure, resulting in non-self-averaging long-range domain correlations.
What carries the argument
Charge ordering within the polar head aggregates of the alcohol models, which influences mixing and creates micro heterogeneous structures.
If this is right
- Short and long alcohols remain well mixed in 2D at all proportions instead of demixing.
- Ideality competes with micro phase separation inside the aggregates of polar heads.
- Domain correlations exhibit non-self-averaging behavior in the long-range part.
- Mixtures of associating molecules follow special fluctuation rules not conventional in 3D.
Where Pith is reading between the lines
- Charge ordering could be a general mechanism stabilizing mixtures in low-dimensional systems.
- Experimental 2D films of alcohol mixtures might show these ideal mixing properties.
- The non-self-averaging correlations may affect thermodynamic properties like compressibility in 2D.
- Similar behaviors could appear in other 2D hydrogen-bonding fluids.
Load-bearing premise
The site-based models capture the essential charge distributions and interactions of real alcohols sufficiently to determine the mixing behavior in two dimensions.
What would settle it
If simulations with more detailed molecular models or experiments on 2D alcohol layers show macroscopic demixing between short and long chains, the role of charge ordering in preventing separation would be questioned.
Figures











read the original abstract
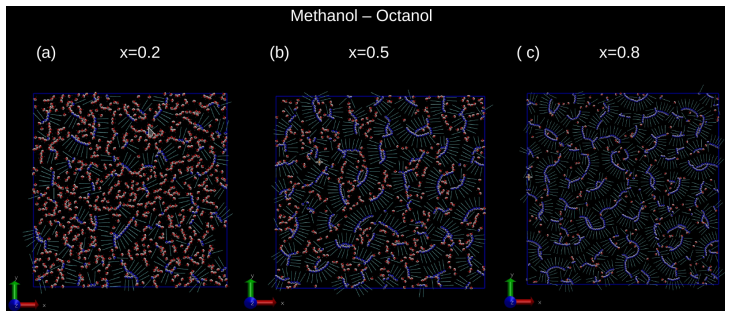
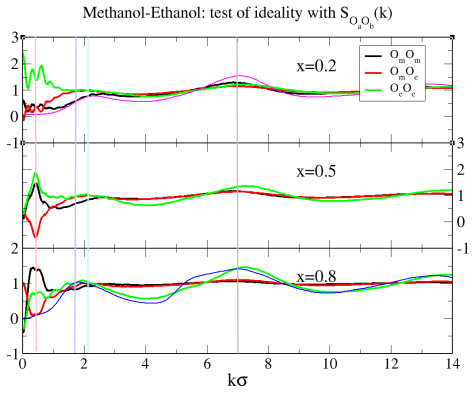
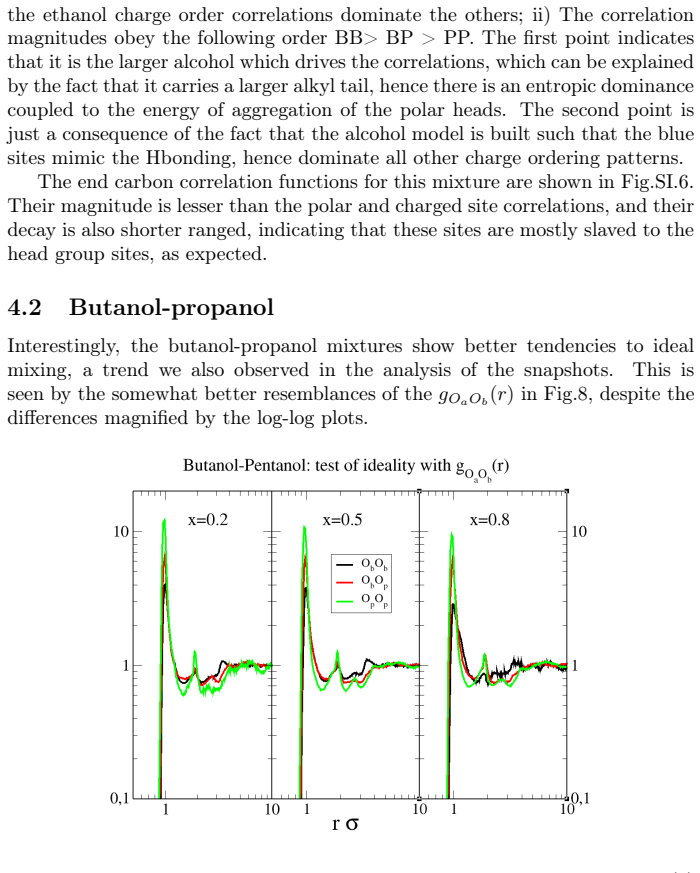
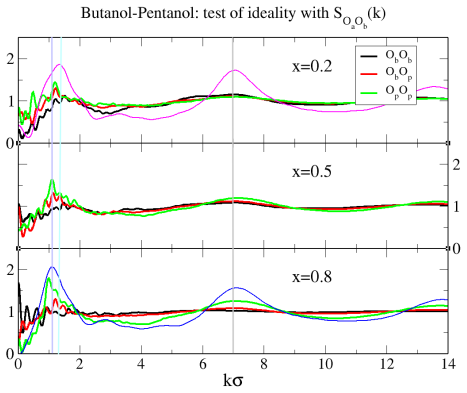
Binary mixtures of two dimensional, site-based models of alcohols are investigated by computer simulations, with a focus on ideal mixing, local clustering and miscibility trends. Four representative systems are considered: methanol/ethanol, butanol/pentanol, methanol/pentanol, and methanol/octanol. The models retain chemical specificity, while allowing to investigate dimensional constraints and uncover non/trivial micro/structurations. Two unexpected results are observed. First, mixtures of short and long alcohols are well mixed, instead of the macroscopic phase separation found in their three-dimensional counterparts. Second, ideality and micro phase separation compete within the chain like polar head aggregates. These behaviors cannot be explained solely by enhanced fluctuations in two dimensions, and instead point to a key role of charge ordering in shaping the local structure. The resulting interplay between concentration fluctuations and micro heterogeneous aggregation is analyzed through snapshots, site/site distribution functions, structure factors and Kirkwood Buff integrals. In particular, the analysis reveals that the domain correlations in the long range part of the correlations have an intriguing non self averaging behaviour, similar to that found in the real systems, indicating that mixtures of associating molecules are not ruled by conventional fluctuations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports molecular dynamics simulations of four binary mixtures of 2D site-based alcohol models (methanol/ethanol, butanol/pentanol, methanol/pentanol, methanol/octanol). It finds ideal mixing for short/long chain pairs (contrary to macroscopic demixing in 3D analogs), competition between ideality and micro-phase separation within polar-head aggregates, and non-self-averaging long-range domain correlations in Kirkwood-Buff integrals and structure factors. These are interpreted as arising from charge ordering within the head-group aggregates rather than from 2D fluctuations alone.
Significance. If the central interpretation holds, the work identifies a distinctive role for charge ordering in suppressing macroscopic demixing and producing non-self-averaging correlations in 2D associating fluids, with potential relevance to surface monolayers and 2D materials. The direct comparison to 3D behavior and the use of chemically specific site models are strengths; the reported non-self-averaging behavior, if robustly quantified, would be a notable addition to the literature on 2D mixtures.
major comments (2)
- [Abstract and discussion] Abstract and discussion sections: the claim that the observed ideal mixing and absence of demixing 'cannot be explained solely by enhanced fluctuations in two dimensions, and instead point to a key role of charge ordering' is load-bearing for the paper's interpretation, yet all reported simulations retain the partial charges of the site-based models. No control runs with electrostatic interactions disabled (while retaining Lennard-Jones and bonding parameters) are described, leaving open whether 2D geometry and hydrogen-bond geometry alone produce the same mixing behavior.
- [Results (Kirkwood-Buff and structure-factor analysis)] Results section on Kirkwood-Buff integrals and structure factors: the non-self-averaging long-range domain correlations are presented as a central finding, but the manuscript provides no quantitative error bars, block-averaging statistics, or finite-size scaling analysis to establish that the observed non-self-averaging is statistically significant rather than an artifact of limited sampling or system size.
minor comments (2)
- [Abstract] The abstract states that the models 'retain chemical specificity' but does not list the explicit partial-charge values or Lennard-Jones parameters used; these should be tabulated for reproducibility.
- [Figures] Figure captions for snapshots and RDFs should explicitly state the simulation temperature, pressure, and total number of molecules to allow direct comparison with the 3D literature.
Simulated Author's Rebuttal
We thank the referee for the careful reading of our manuscript and the constructive comments, which have helped clarify several important aspects. We address each major comment below and outline the revisions we will make.
read point-by-point responses
-
Referee: [Abstract and discussion] Abstract and discussion sections: the claim that the observed ideal mixing and absence of demixing 'cannot be explained solely by enhanced fluctuations in two dimensions, and instead point to a key role of charge ordering' is load-bearing for the paper's interpretation, yet all reported simulations retain the partial charges of the site-based models. No control runs with electrostatic interactions disabled (while retaining Lennard-Jones and bonding parameters) are described, leaving open whether 2D geometry and hydrogen-bond geometry alone produce the same mixing behavior.
Authors: We agree that the referee has identified a key limitation in the current evidence for our central interpretation. The claim regarding charge ordering is indeed central, and the absence of control simulations with partial charges set to zero leaves the role of electrostatics versus purely geometric 2D effects incompletely tested. In the revised manuscript we will add results from such control simulations (with electrostatic interactions disabled while retaining Lennard-Jones and bonding parameters). These will be used to directly compare mixing behavior, local structure, and Kirkwood-Buff integrals, allowing us to quantify the contribution of charge ordering. The abstract and discussion will be updated to reflect the new findings and to qualify the original statement accordingly. revision: yes
-
Referee: [Results (Kirkwood-Buff and structure-factor analysis)] Results section on Kirkwood-Buff integrals and structure factors: the non-self-averaging long-range domain correlations are presented as a central finding, but the manuscript provides no quantitative error bars, block-averaging statistics, or finite-size scaling analysis to establish that the observed non-self-averaging is statistically significant rather than an artifact of limited sampling or system size.
Authors: The referee correctly notes that the statistical robustness of the reported non-self-averaging behavior requires more explicit quantification. While the original analysis already examined trends across multiple compositions and system sizes, we did not provide block-averaged error estimates or a dedicated finite-size scaling study. In the revised manuscript we will add quantitative error bars obtained from block averaging over independent trajectories, together with a finite-size scaling analysis for the long-range part of the Kirkwood-Buff integrals and structure factors. This will establish that the non-self-averaging persists with increasing system size and is not an artifact of sampling limitations. revision: yes
Circularity Check
No circularity detected; results from direct simulation
full rationale
The paper reports outcomes from explicit molecular dynamics simulations of site-based 2D alcohol models, with mixing trends, domain correlations, and non-self-averaging behavior extracted directly from computed RDFs, structure factors, and Kirkwood-Buff integrals. No equations or parameters are fitted to the target observations, no self-definitional loops exist, and no load-bearing claims reduce to self-citations or ansatzes that presuppose the reported results. Comparisons to 3D demixing and attribution to charge ordering are interpretive inferences grounded in the model setup and external benchmarks, leaving the derivation chain independent and self-contained.
Axiom & Free-Parameter Ledger
axioms (2)
- standard math Standard assumptions of statistical mechanics for molecular simulations (ergodicity, adequate sampling of phase space)
- domain assumption The site-based models retain sufficient chemical specificity to represent real alcohol behavior in 2D
Forward citations
Cited by 1 Pith paper
-
Aqueous-alcohol mixtures in dimension two: miscibility and micro-segregation
2D water-alcohol mixtures stay fully miscible with micro-segregation that grows with alcohol chain length, driven by water self-aggregation but limited by rim contacts, due to reorganization of fluctuations under char...
Reference graph
Works this paper leans on
-
[1]
Rowlinson, J. S.; Swinton, F. L. Liquids and Liquids Mixtures. Butter- worths Scientific, 3rd ed., 1982
work page 1982
-
[2]
Atkins, P.; de Paula, J. Atkins Physical Chemistry. Oxford UniversityPress, London, 2010
work page 2010
-
[3]
Po z ar, M.; Lovrin c evi\' c , B.; Zorani\' c , L.; Mijakovi\' c , M.; Sokoli\' c , F.; Perera, A. A re-appraisal of the concept of ideal mixtures through a computer simulation study of the methanol-ethanol mixtures. The Journal of Chemical Physics 2016. 145, 064509
work page 2016
- [4]
-
[5]
Letcher, T. M.; Raal, J. D. Binary systems of nonelectrolytes containing alcohols (c1--c9). Journal of Physical and Chemical Reference Data 2003. 32, 1065--1357
work page 2003
-
[6]
From solutions to molecular emulsions
Perera, A. From solutions to molecular emulsions. Pure and Applied Chemistry 2016. 88, 189--206
work page 2016
-
[7]
Lum, K.; Chandler, D.; Weeks, J. D. Hydrophobicity at small and large length scales. Journal of Physical Chemistry 1999. 103, 4570 -- 4577
work page 1999
-
[8]
Pratt, L. R.; Chandler, D. Theory of the hydrophobic effect. The Journal of Chemical Physics 1977. 67, 3683
work page 1977
-
[9]
Willard, A. P.; Chandler, D. The role of solvent fluctuations in hydrophobic assembly. Journal of Physical Chemistry 2008. 112, 6187--6192
work page 2008
-
[10]
Hydrophobic ambivalence: Teetering on the edge of randomness
Ben-Amotz, D. Hydrophobic ambivalence: Teetering on the edge of randomness. The Journal of Physical Chemistry Letters 2015. 6, 1696--1701
work page 2015
-
[11]
Comment on <the mechanism of hydrophobic solvation depends on solute radius> j
Graziano, G. Comment on <the mechanism of hydrophobic solvation depends on solute radius> j. phys.chem. b 2000, 104, 1326. Journal of Physical Chemistry B 2001. 105, 2079 -- 2081
work page 2000
-
[12]
On the intactness of hydrogen bonds around nonpolar solutes dissolved in water
Graziano, G.; Lee, B. On the intactness of hydrogen bonds around nonpolar solutes dissolved in water. Journal of Physical Chemistry B 2005. 109, 8103 -- 8107
work page 2005
-
[13]
S Weerasinghe, P. E. S. Kirkwood-buff derived force field for mixtures of acetone and water. The Journal of chemical physics 2003. 118, 10663
work page 2003
-
[14]
Chitra, R.; Smith, P. E. Properties of 2,2,2-trifluoroethanol and water mixtures. The Journal of Chemical Physics 2001. 114, 426
work page 2001
-
[15]
Lee, M. E.; van der Vegt, N. F. A. A new force field for atomistic simulations of aqueoustertiary butanol solutions. The Journal of Chemical Physics 2005. 122, 114509
work page 2005
-
[16]
Mountain, R. D. Microstructure and hydrogen bonding in water-acetonitrile mixtures. Journal of Physical Chemistry B 2010. 114, 16460 -- 16464
work page 2010
-
[17]
Dixit, S.; Crain, J.; Poon, W. C. K.; Finney, J. L.; Soper, A. K. Molecular segregation observed in a concentrated alcohol--water solution. Nature 2002. 416, 829--832
work page 2002
-
[18]
Bunkin, N. F.; Shkirin, A. V.; Lyakhov, G. A.; Kobelev, A. V.; Penkovi, N. V.; Ugraitskaya, S. V.; Fesenko, E. E. Droplet-like heterogeneity of aqueous tetrahydrofuran solutions at thesubmicrometer scale. The Journal of Chemical Physics 2016. 145, 184501
work page 2016
-
[19]
Sedl\' a k, M.; Rak, D. On the origin of mesoscale structures in aqueous solutions of tertiary butyl alcohol: The mystery resolved. The Journal of Physical Chemistry B 2014. 118, 2726--2737. PMID: 24559045
work page 2014
-
[20]
Camel back shaped kirkwood - buff integrals
Perera, A.; Po z ar, M.; Lovrin c evi\' c , B. Camel back shaped kirkwood - buff integrals. The Journal of Chemical Physics 2022. 156, 124503
work page 2022
-
[21]
Molecular emulsions: from charge order to domain order
Perera, A. Molecular emulsions: from charge order to domain order. Phys. Chem. Chem. Phys. 2017. 19, 28275--28285
work page 2017
-
[22]
Mesoscopic correlations in aqueous alkylaminemixtures between molecular and micro emulsions
Perera, A. Mesoscopic correlations in aqueous alkylaminemixtures between molecular and micro emulsions. Physics of fluids 2026. 38, 017125
work page 2026
-
[23]
Dong Nam ShinJan, B. F. N. E., W. WijnenJan; Wakisaka, A. On the origin of microheterogeneity: A mass spectrometric study of dimethylsulfoxide-water binary mixture. Journal of Physical Chemistry 2001. 105, 6759--6762
work page 2001
-
[24]
Site-site interaction model for alcohol models in two-dimension
Perera, A. Site-site interaction model for alcohol models in two-dimension. Journal of Molecular Liquids 2025. 422, 126944
work page 2025
-
[25]
Tom s i c , M.; Jamnik, A.; Fritz-Popovski, G.; Glatter, O.; Vl c ek, L. Structural properties of pure simple alcohols from ethanol, propanol, butanol, pentanol, to hexanol: Comparing monte carlo simulations with experimental saxs data. The Journal of Physical Chemistry B 2007. 111, 1738--1751
work page 2007
-
[26]
Po z ar, M.; Bolle, J.; Sternemann, C.; Perera, A. On the x-ray scattering pre-peak of linear mono-ols and the related microstructure from computer simulations. The Journal of Physical Chemistry B 2020. 124, 8358--8371
work page 2020
-
[27]
Simple two-dimensional models of alcohols
Papez, P.; Urbic, T. Simple two-dimensional models of alcohols. Phys. Rev. E 2022. 105, 054608
work page 2022
-
[28]
Statistical Mechanics of Waterlike Particles in Two Dimensions
Ben Naim, A. Statistical Mechanics of Waterlike Particles in Two Dimensions. I. Physical Model and Application of the Percus - Yevick Equation . The Journal of Chemical Physics 1971. 54, 3682--3695
work page 1971
-
[29]
Brini, E.; Fennell, C. J.; Serra, M. F.; HribarLee, B.; Luk s i\' c , M.; Dill, K. A. How water properties are encoded in its molecular structure and energies. Chemical Reviews 2017. 117, 12385--12414. PMID: 28949513
work page 2017
-
[30]
Ogrin, P.; Urbic, T. Angle-dependent integral equation theory improves results of thermodynamics and structure of rose water model. The Journalof Chemical Physics 2023. 159, 114505
work page 2023
-
[31]
A site-site interaction two-dimensional model with water like structural properties
Bar\' e , T.; Besserve, M.; Urbic, T.; Perera, A. A site-site interaction two-dimensional model with water like structural properties. Journal of Molecular Liquids 2023. 386, 122475
work page 2023
-
[32]
The statistical mechanical theory of solutions
Kirkwood, J.; Buff, F. The statistical mechanical theory of solutions. i. The Journal of Chemical Physics 1951. 19, 774
work page 1951
-
[33]
A Guide to Monte Carlo Simulations in Statistical Physics
Binder, K.; Stauffer, D. A Guide to Monte Carlo Simulations in Statistical Physics. Cambridge University Press, 1997
work page 1997
-
[34]
Harris, A. B. Effect of random defects on the critical behaviour of ising models. Journal of Physics C: Solid State Physics 1974. 7, 1671--1692
work page 1974
-
[35]
Lack of self-averaging in critical disordered systems
Wiseman, I.; Domany, E. Lack of self-averaging in critical disordered systems. Physical Review Letters 1998. 81, 22--25
work page 1998
-
[36]
The influence of charge ordering in the microscopic structure of monohydroxy alcohols
Po z ar, M.; Lovrin c evi\' c , B.; Perera, A. The influence of charge ordering in the microscopic structure of monohydroxy alcohols. Journal of Physics: Condensed Matter 2024. 36, 265102
work page 2024
-
[37]
Ganguly, P.; van der Vegt, N. F. A. Convergence of sampling kirkwood-buff integrals of aqueous solutions with molecular dynamics simulations. Journal of Chemical Theory and Computation 2013. 9, 1347 -- 1355
work page 2013
-
[38]
K.; Bedeaux, D.; Kjelstrup, S.; Vlugt, T
Kr\" u ger, P.; Schnell, S. K.; Bedeaux, D.; Kjelstrup, S.; Vlugt, T. J. H.; Simon, J.-M. Kirkwood-buff integrals for finite volumes. Journal of Physical Chemistry Letters 2013. 4, 235 -- 238
work page 2013
-
[39]
Dednam, W.; Botha, A. E. Evaluation of kirkwood-buff integrals via finite sizescaling: a large scale molecular dynamics study. Journal of Physics: Conference Series 2015. 574, 012092
work page 2015
-
[40]
Methanol-ethanol ideal mixtures as a test ground for the computation of kirkwood-buff integrals
Lovrin c evi\' c , B.; Bella, A.; Tenoux-Rachidi, L.; I., M., Po z ar; Sokoli\' c , F.; Perera, A. Methanol-ethanol ideal mixtures as a test ground for the computation of kirkwood-buff integrals. Journal of Molecular Liquids 2019. 293, 111447
work page 2019
-
[41]
Gennes, P. G. D.; Prost, J. The Physics of Liquid Crystals. Oxford University Press, 1993
work page 1993
-
[42]
Principles of Condensed Matter Physics
Chaikin, P.; Lubensky, T. Principles of Condensed Matter Physics. Cambridge University Press, 2000
work page 2000
-
[43]
, " * write output.state after.block = add.period write newline
ENTRY address author booktitle chapter edition editor eid howpublished institution journal key month note number organization pages publisher school series title type volume year label INTEGERS output.state before.all mid.sentence after.sentence after.block FUNCTION init.state.consts #0 'before.all := #1 'mid.sentence := #2 'after.sentence := #3 'after.bl...
-
[44]
" write newline "" before.all 'output.state := FUNCTION n.dashify 't := "" t empty not t #1 #1 substring "-" = t #1 #2 substring "--" = not "--" * t #2 global.max substring 't := t #1 #1 substring "-" = "-" * t #2 global.max substring 't := while if t #1 #1 substring * t #2 global.max substring 't := if while FUNCTION word.in bbl.in capitalize " " * FUNCT...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.