Recognition: unknown
Advancing Ligand-based Virtual Screening and Molecular Generation with Pretrained Molecular Embedding Distance
Pith reviewed 2026-05-08 04:00 UTC · model grok-4.3
The pith
Pretrained embedding distances serve as an effective training-free measure of molecular similarity for virtual screening and generation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Pretrained embedding distance (PED) is obtained directly from the latent representations of pretrained molecular models without any task-specific training or additional data curation. The distance metric exhibits distinct correlations with conventional similarity measures such as fingerprint-based Tanimoto coefficients and 3D shape overlays. When applied to ranking, PED identifies relevant molecules for virtual screening; when used in reward design, it directs generative models toward structurally appropriate outputs. These results indicate that embeddings from general pretrained models already contain sufficient structural information to support similarity-aware tasks in ligand-based drug设计
What carries the argument
Pretrained embedding distance (PED), the distance computed between molecular embeddings produced by general-purpose pretrained encoders, which supplies a similarity signal without hand-crafted descriptors or supervised fine-tuning.
If this is right
- PED ranks candidate molecules for virtual screening at scale using only forward passes through existing pretrained models.
- PED can be inserted as a reward term in molecular generative models to bias outputs toward structural analogs without extra supervision.
- Distinct correlations between PED and traditional metrics imply that PED captures complementary aspects of molecular similarity.
- Pretrained embeddings encode rich enough structural detail to replace or augment hand-crafted descriptors in multiple ligand-based workflows.
Where Pith is reading between the lines
- If PED maintains performance across broader chemical libraries, it could support similarity searches in compound collections too large for 3D overlay methods.
- The training-free nature suggests PED might enable rapid similarity assessment for entirely new targets where labeled data is unavailable.
- Combining PED with existing generative pipelines could provide an efficient way to explore analog series while controlling for structural similarity.
Load-bearing premise
Embeddings from general pretrained molecular models already contain the structural information required to measure similarity effectively across different drug targets without any task-specific training or data curation.
What would settle it
A virtual screening benchmark on a target where PED-based rankings of known actives versus decoys show substantially lower enrichment than Tanimoto or shape-based rankings.
Figures




read the original abstract
Molecular similarity plays a central role in ligand-based drug discovery, such as virtual screening, analog searching, and goal-directed molecular generation. However, traditional similarity measures, ranging from fingerprint-based Tanimoto coefficients to 3D shape overlays, are often computationally expensive at scale or rely on hand-crafted molecular descriptors. Meanwhile, many deep learning approaches to similarity-aware design still depend on similarity-specific supervision or costly data curation, limiting their generality across targets. In this work, we propose pretrained embedding distance (PED) as an effective alternative, computed directly from pretrained molecular models without task-specific training. Experimental results show that PED exhibits distinct correlations with traditional similarity metrics, and performs effectively in both ranking molecules for virtual screening and guiding molecular generation via reward design. These findings suggest that pretrained molecular embeddings capture rich structural information and can serve as a promising and scalable similarity measurement for modern AI-aided drug discovery.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes pretrained embedding distance (PED) as a similarity metric computed directly from embeddings of general pretrained molecular models (no task-specific fine-tuning or supervision). It claims that PED exhibits distinct correlations with traditional measures such as Tanimoto coefficients, performs effectively for ranking molecules in ligand-based virtual screening, and can serve as a reward signal to guide goal-directed molecular generation.
Significance. If the empirical claims hold with proper validation, PED would offer a scalable, low-supervision alternative to hand-crafted descriptors or supervised similarity models, leveraging existing pretrained molecular representations for both screening and generation tasks. A notable strength is the parameter-free derivation from off-the-shelf models, which aligns with efforts to reduce task-specific data curation in AI-aided drug discovery.
major comments (2)
- [Experimental validation and results sections] The central claim that PED 'performs effectively' in virtual screening and generation without task-specific supervision rests on an untested assumption that general pretraining corpora (e.g., broad ZINC/ChEMBL sets) already encode the structural signals needed for arbitrary downstream targets. No cross-target hold-out experiments, ablation on pretraining-data overlap, or analysis of target classes underrepresented in pretraining are reported; this is load-bearing for the generality assertion.
- [Abstract and Experimental Results] Abstract and results sections assert 'distinct correlations' and 'effective' performance but supply no quantitative metrics (e.g., Spearman/Pearson coefficients, AUC-ROC or enrichment factors for screening, success rates or property improvements for generation), baselines, error bars, dataset sizes, or validation protocols. Without these, the evidence for the effectiveness claim cannot be evaluated.
minor comments (2)
- [Methods] Clarify the exact pretrained models used (architecture, training corpus, embedding dimension) and the precise distance function (e.g., Euclidean, cosine) in the Methods section to ensure reproducibility.
- [Abstract] The abstract would be strengthened by including one or two key quantitative highlights (with numbers) rather than qualitative statements only.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address each major comment point by point below, indicating where revisions will be made to strengthen the presentation and validation of our claims.
read point-by-point responses
-
Referee: [Experimental validation and results sections] The central claim that PED 'performs effectively' in virtual screening and generation without task-specific supervision rests on an untested assumption that general pretraining corpora (e.g., broad ZINC/ChEMBL sets) already encode the structural signals needed for arbitrary downstream targets. No cross-target hold-out experiments, ablation on pretraining-data overlap, or analysis of target classes underrepresented in pretraining are reported; this is load-bearing for the generality assertion.
Authors: We agree that the generality of PED across arbitrary targets is a key aspect of our claims and that the current experiments do not include explicit cross-target hold-out validation, ablations on pretraining-data overlap, or targeted analysis of underrepresented target classes. Our reported results rely on standard benchmarks drawn from diverse targets, but these do not fully isolate the effects of pretraining data composition. In the revised manuscript we will add a dedicated subsection with cross-target hold-out experiments, an ablation study varying the degree of pretraining data overlap with evaluation targets, and a discussion of performance on target classes with limited representation in the pretraining corpus. revision: yes
-
Referee: [Abstract and Experimental Results] Abstract and results sections assert 'distinct correlations' and 'effective' performance but supply no quantitative metrics (e.g., Spearman/Pearson coefficients, AUC-ROC or enrichment factors for screening, success rates or property improvements for generation), baselines, error bars, dataset sizes, or validation protocols. Without these, the evidence for the effectiveness claim cannot be evaluated.
Authors: We acknowledge that the abstract and main text do not explicitly report the requested quantitative metrics, baselines, error bars, dataset sizes, or detailed validation protocols, even though the experimental sections contain supporting figures and comparisons. This omission makes independent evaluation of the claims difficult. In the revised version we will expand the abstract to include key quantitative results (e.g., correlation coefficients, AUC-ROC, enrichment factors, and generation success rates), add a summary table with all metrics, error bars, and statistical details, clearly list dataset sizes and validation protocols, and ensure all baselines are described with the same rigor. revision: yes
Circularity Check
No circularity: PED defined directly from pretrained models with independent experimental validation
full rationale
The paper defines pretrained embedding distance (PED) explicitly as a distance metric computed from embeddings of general pretrained molecular models, with no task-specific training, fitting, or supervision mentioned. Performance claims rest on separate experimental evaluations for virtual screening ranking and reward-guided generation, which are downstream tests rather than quantities derived by construction from the definition itself. No self-citations, uniqueness theorems, or ansatzes are invoked to justify the core method; the abstract and described approach remain self-contained against external benchmarks without reducing predictions to fitted inputs or self-referential steps.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URL https://www.pnas.org/doi/abs/10.1073/ pnas.2415665122
doi: 10.1073/pnas.2415665122. URL https://www.pnas.org/doi/abs/10.1073/ pnas.2415665122. Oleksandr O. Grygorenko, Dmytro S. Radchenko, Igor Dziuba, Alexander Chuprina, Kateryna E. Gubina, and Yurii S. Moroz. Generating multibillion chemical space of readily accessible screen- ing compounds.iScience, 23(11):101681, 2020. ISSN 2589-0042. doi: https://doi.or...
-
[2]
doi: 10.1038/s41598-025-99785-0
ISSN 2045-2322. doi: 10.1038/s41598-025-99785-0. URL https://doi.org/10.1038/ s41598-025-99785-0. Paul C. D. Hawkins, A. Geoffrey Skillman, and Anthony Nicholls. Comparison of shape-matching and docking as virtual screening tools.Journal of Medicinal Chemistry, 50(1):74–82, 2007. doi: 10.1021/jm0603365. URLhttps://doi.org/10.1021/jm0603365. PMID: 17201411...
-
[3]
URL https://doi.org/10.1021/acs.jcim.0c01015
doi: 10.1021/acs.jcim.0c01015. URL https://doi.org/10.1021/acs.jcim.0c01015. PMID: 33301333. Cheng-Han Li and Daniel P. Tabor. Generative organic electronic molecular design informed by quantum chemistry.Chem. Sci., 14:11045–11055, 2023. doi: 10.1039/D3SC03781A. URL http://dx.doi.org/10.1039/D3SC03781A. Xinhao Li and Denis Fourches. Inductive transfer lea...
-
[4]
doi: 10.3390/molecules28114430
ISSN 1420-3049. doi: 10.3390/molecules28114430. URL https://www.mdpi.com/ 1420-3049/28/11/4430. Yifei Wang, Yunrui Li, Lin Liu, Pengyu Hong, and Hao Xu. Advancing drug discovery with enhanced chemical understanding via asymmetric contrastive multimodal learning.Journal of Chemical Information and Modeling, 65(13):6547–6557, 2025a. doi: 10.1021/acs.jcim.5c...
-
[5]
Smiles, a chemical language and information system
doi: 10.1021/ci00057a005. URLhttps://doi.org/10.1021/ci00057a005. Ronald J. Williams. Simple statistical gradient-following algorithms for connectionist rein- forcement learning.Machine Learning, 8(3):229–256, May 1992. ISSN 1573-0565. doi: 10.1007/BF00992696. URLhttps://doi.org/10.1007/BF00992696. Bohao Xu, Yingzhou Lu, Chenhao Li, Ling Yue, Xiao Wang, T...
-
[6]
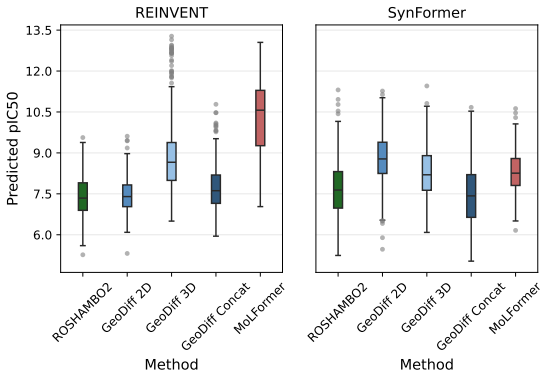
models as two representative pretrained molecular models to compute PED. GeoDiff was pretrained on the GEOM-Drugs Axelrod and Gómez-Bombarelli [2022] dataset, and MoLFormer was the XL version (https://huggingface.co/ibm-research/MoLFormer-XL-both-10pct ) pretrained on 1.1B molecules from ZINC and PubChem (10% of both datasets). Both 2D GIN and 3D SchNet e...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.