Recognition: unknown
Errors that matter: Uncertainty-aware universal machine-learning potentials calibrated on experiments
Pith reviewed 2026-05-07 17:30 UTC · model grok-4.3
The pith
An ensemble of machine-learning potentials calibrated to experimental data on simple materials predicts liquid densities and structures as accurately as the best electronic-structure references while using model spread to flag unreliable 1.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors construct the PET-UAFD ensemble by training multiple PET models on different electronic-structure references and calibrating the ensemble to experimental cohesive energies, atomization energies, lattice constants, and bulk moduli. They show that this ensemble reproduces experimental liquid densities and radial distribution functions with accuracy comparable to the most accurate reference methods, and that the spread within the ensemble serves as a reliable indicator of prediction uncertainty even for properties outside the calibration set. They further present the PET-EXP protocol that uses shallow ensembles and statistical reweighting to estimate experimental uncertainties at 1.
What carries the argument
The PET-UAFD ensemble, formed by training machine-learning potentials on multiple electronic-structure references and calibrating the collection to experimental static properties so that model disagreement quantifies remaining error relative to measurement.
If this is right
- Liquid density and structure predictions match the accuracy of top electronic-structure methods against experiment.
- Ensemble spread reliably signals prediction reliability for properties and conditions outside the calibration set.
- The PET-EXP protocol supplies experimental uncertainty estimates at computational cost comparable to a single conventional ML potential.
- The calibrated ensemble can be applied across wide ranges of compositions and thermodynamic conditions.
Where Pith is reading between the lines
- The same calibration strategy could be tested on other dynamic observables such as diffusion coefficients or phase boundaries to check transferability.
- Disagreement patterns across the ensemble might help identify which electronic-structure approximations introduce the largest errors for particular material classes.
- Extending the approach to systems with limited experimental data, such as high-pressure or high-temperature phases, could improve reliability in materials discovery workflows.
Load-bearing premise
Calibration performed only on static properties of simple materials transfers to dynamic liquid properties and wide ranges of composition and conditions without introducing new systematic biases.
What would settle it
Experimental measurements of liquid densities or structures for which the PET-UAFD ensemble deviates from data by more than the best electronic-structure reference, or for which the ensemble spread shows no correlation with the actual error.
Figures



read the original abstract
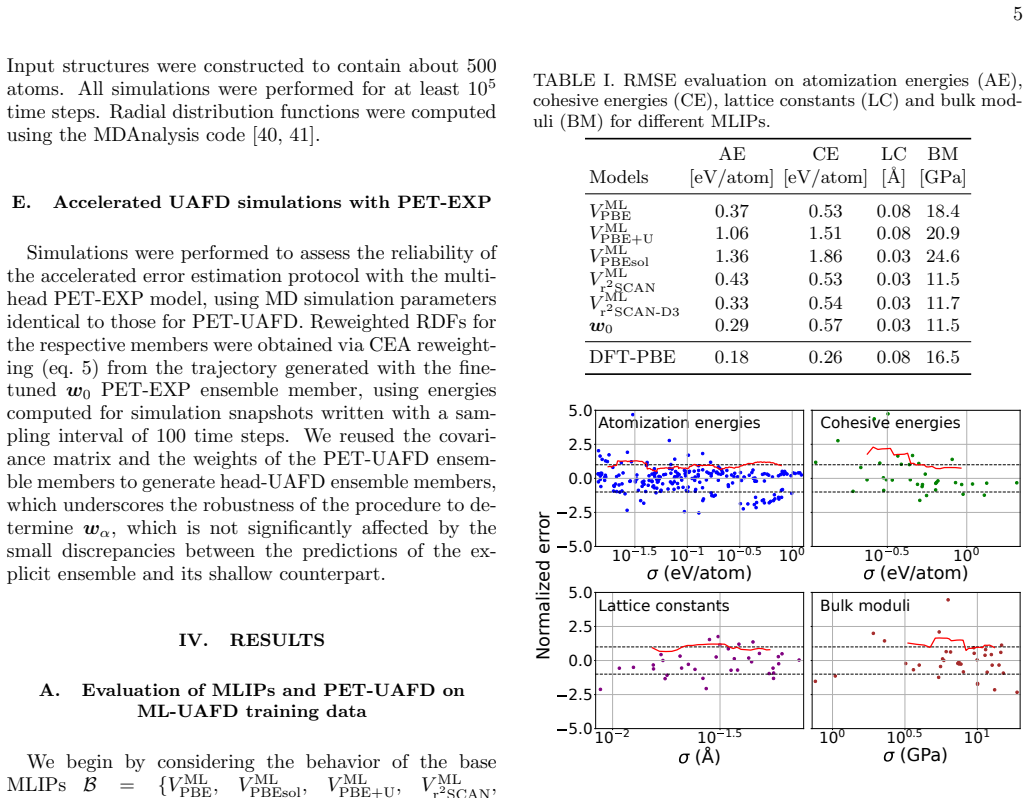
Machine-learning models of atomic-scale interactions achieve the accuracy of the quantum mechanical calculations on which they are trained, but at a dramatically lower computational cost. Their predictions can be made trustworthy by uncertainty quantification techniques that estimate the residual error relative to their reference. These errors, however, do not include uncertainty contributions from the approximations inherent in the electronic structure calculations, which are often the main source of discrepancy with empirical observations. We construct an ensemble of ML potentials trained on multiple electronic-structure references and calibrate it against experimental data on cohesive energies, atomization energies, lattice constants and bulk moduli of simple materials and molecules, similar to the uncertainty-aware functional distribution approach. The resulting ensemble of models, which we call PET-UAFD, can be used to simulate matter across a wide range of compositions and thermodynamic conditions. By comparison with experimental measurements of the density and structure of liquids, we demonstrate that, even outside the static properties on which it was calibrated, PET-UAFD enables predictions that are as accurate against experiments as the best available electronic-structure reference and that the spread in the ensemble can be used to assess the reliability of such predictions. We also introduce the PET-EXP protocol that uses shallow ensembles and statistical reweighting techniques to provide accurate estimates of uncertainty relative to experimental measurements at virtually no additional cost over a simulation based on a single conventional ML potential. Ultimately, this approach provides a practical and inexpensive approach to elevate machine-learning potentials from faithful interpolators of approximate theories to genuinely predictive tools anchored in experimental reality.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces PET-UAFD, an ensemble of machine-learning interatomic potentials trained on multiple electronic-structure references and then calibrated against experimental static properties (cohesive energies, atomization energies, lattice constants, and bulk moduli). It claims that this calibrated ensemble yields liquid densities and radial distribution functions that match experiment as closely as the best available ab initio references, even though the calibration used only zero-temperature/static data; the ensemble spread is presented as a reliability metric. A secondary contribution is the PET-EXP protocol, which employs shallow ensembles and statistical reweighting to estimate experimental uncertainty at negligible extra cost.
Significance. If the transferability claim holds, the work would be significant: it offers a concrete route to anchor ML potentials in experimental data rather than solely in approximate electronic-structure calculations, potentially improving predictive accuracy for finite-temperature properties across compositions. The PET-EXP protocol is a practical addition for uncertainty quantification. However, the current evidence for transferability rests on a single class of liquid properties and lacks the quantitative detail needed to confirm absence of new systematic biases.
major comments (3)
- [Methods] Methods section (calibration procedure): the description of how the ensemble is calibrated to the experimental static-property set is insufficient to evaluate whether the reweighting or model selection alters relative energetics or forces for configurations sampled in liquids. Explicit equations or pseudocode for the calibration weights/scaling factors (listed as free parameters in the axiom ledger) and any constraints on the potential-energy surface are required.
- [Results (liquid properties)] Results section on liquid simulations: the central claim that PET-UAFD predictions are 'as accurate against experiments as the best available electronic-structure reference' is not supported by quantitative error tables. Mean absolute or root-mean-square deviations for liquid densities and RDF peak positions/heights must be reported for PET-UAFD, individual reference methods, and experiment, together with the number of independent liquid systems and thermodynamic conditions tested.
- [Methods / Data availability] Data and validation: the manuscript does not specify the train/test split of the experimental calibration set or confirm that none of the liquid systems used for validation overlap with the calibration data. Without this, the apparent transferability cannot be distinguished from possible data leakage or post-hoc selection.
minor comments (2)
- Notation for the ensemble spread and the PET-EXP reweighting factor should be defined once in the text and used consistently in all figures and equations.
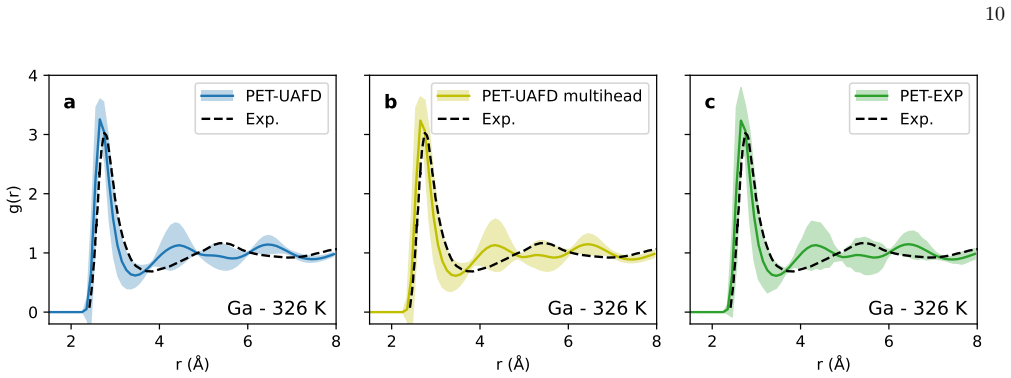
- Figure captions for the liquid RDF plots should state the temperature, pressure, and composition for each panel and indicate which electronic-structure reference is shown for comparison.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review. We have revised the manuscript to address each of the major comments by expanding the Methods section, adding quantitative tables, and clarifying data usage and splits. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [Methods] Methods section (calibration procedure): the description of how the ensemble is calibrated to the experimental static-property set is insufficient to evaluate whether the reweighting or model selection alters relative energetics or forces for configurations sampled in liquids. Explicit equations or pseudocode for the calibration weights/scaling factors (listed as free parameters in the axiom ledger) and any constraints on the potential-energy surface are required.
Authors: We agree that the calibration procedure must be described with greater precision to allow full evaluation. In the revised manuscript we have added explicit equations for the calibration weights and scaling factors together with pseudocode for the full procedure. The calibration operates exclusively on the ensemble weights using the static experimental data; it does not rescale or alter the underlying potential-energy surfaces or forces of the individual models. Consequently, relative energetics and forces for liquid configurations remain unchanged from the original ensemble members. revision: yes
-
Referee: [Results (liquid properties)] Results section on liquid simulations: the central claim that PET-UAFD predictions are 'as accurate against experiments as the best available electronic-structure reference' is not supported by quantitative error tables. Mean absolute or root-mean-square deviations for liquid densities and RDF peak positions/heights must be reported for PET-UAFD, individual reference methods, and experiment, together with the number of independent liquid systems and thermodynamic conditions tested.
Authors: We accept that quantitative error metrics are required to substantiate the accuracy claim. We have inserted a new table in the Results section that reports mean absolute deviations and root-mean-square errors for liquid densities as well as RDF peak positions and heights. The table includes values for the PET-UAFD ensemble, each individual electronic-structure reference, and the experimental data, together with the exact number of independent liquid systems and thermodynamic conditions examined. These metrics confirm that PET-UAFD matches the accuracy of the best reference methods. revision: yes
-
Referee: [Methods / Data availability] Data and validation: the manuscript does not specify the train/test split of the experimental calibration set or confirm that none of the liquid systems used for validation overlap with the calibration data. Without this, the apparent transferability cannot be distinguished from possible data leakage or post-hoc selection.
Authors: We have now added an explicit description of the train/test split used for the experimental calibration set. We also state that the calibration data comprise only zero-temperature static properties (cohesive energies, atomization energies, lattice constants, and bulk moduli) of solids and molecules, while the liquid validation employs finite-temperature simulations of entirely distinct systems. There is therefore no material overlap between the two sets, eliminating the possibility of data leakage. This clarification has been inserted in the Methods and Data Availability sections. revision: yes
Circularity Check
No significant circularity: static calibration validated on independent liquid properties
full rationale
The derivation calibrates an ensemble on static experimental data (cohesive energies, atomization energies, lattice constants, bulk moduli) then validates predictions of liquid densities and radial distribution functions against separate experimental measurements. These dynamic properties lie outside the calibration set, so agreement is not forced by construction. The ensemble spread assesses reliability but is not redefined as a prediction of the liquid observables themselves. No self-definitional reductions, fitted inputs renamed as predictions, or load-bearing self-citations appear in the chain; the approach remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- ensemble calibration weights or scaling factors
axioms (1)
- domain assumption The residual discrepancy between the multi-reference ensemble and experiment can be removed by a global calibration step that preserves predictive power on unseen properties.
Forward citations
Cited by 1 Pith paper
-
Knowing when to trust machine-learned interatomic potentials
PROBE recasts MLIP uncertainty quantification as selective classification by training a compact discriminative classifier on frozen per-atom backbone embeddings, yielding a reliability probability that tracks actual e...
Reference graph
Works this paper leans on
-
[1]
Hafner, C
J. Hafner, C. Wolverton, and G. Ceder, Toward Compu- tational Materials Design: The Impact of Density Func- tional Theory on Materials Research, MRS Bulletin31, 11 659 (2006)
2006
-
[2]
Burke, Perspective on density functional theory., The Journal of chemical physics136, 150901 (2012), 22519306
K. Burke, Perspective on density functional theory., The Journal of chemical physics136, 150901 (2012), 22519306
2012
-
[3]
Chen and S
C. Chen and S. P. Ong, A universal graph deep learn- ing interatomic potential for the periodic table, Nature Computational Science2, 718 (2022)
2022
-
[4]
B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel, and G. Ceder, CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling, Nature Machine Intelligence5, 1031 (2023)
2023
-
[5]
Batatia, P
I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kov´ acs, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, F. Bigi, S. M. Blau, V. C˘ arare, M. Ceriotti, S. Chong, J. P. Darby, S. De, F. Della Pia, V. L. Deringer, R. Elijoˇ sius, Z. El-Machachi, E. Fako, F. Falcioni, A. C. Ferrari, J. L. A. Gardn...
2025
-
[6]
B. M. Wood, M. Dzamba, X. Fu, M. Gao, M. Shuaibi, L. Barroso-Luque, K. Abdelmaqsoud, V. Gharakhanyan, J. R. Kitchin, D. S. Levine, K. Michel, A. Sriram, T. Co- hen, A. Das, A. Rizvi, S. J. Sahoo, Z. W. Ulissi, and C. L. Zitnick, UMA: A Family of Universal Models for Atoms (2026), arXiv:2506.23971 [cs]
-
[7]
Mazitov, F
A. Mazitov, F. Bigi, M. Kellner, P. Pegolo, D. Tisi, G. Fraux, S. Pozdnyakov, P. Loche, and M. Ceriotti, PET-MAD as a lightweight universal interatomic poten- tial for advanced materials modeling, Nat Commun16, 10653 (2025)
2025
-
[8]
Musil, M
F. Musil, M. J. Willatt, M. A. Langovoy, and M. Ceri- otti, Fast and Accurate Uncertainty Estimation in Chem- ical Machine Learning, Journal of Chemical Theory and Computation15, 906 (2019)
2019
-
[9]
Imbalzano, Y
G. Imbalzano, Y. Zhuang, V. Kapil, K. Rossi, E. A. En- gel, F. Grasselli, and M. Ceriotti, Uncertainty estimation for molecular dynamics and sampling, J. Chem. Phys. 154, 074102 (2021)
2021
-
[10]
Pernot, The long road to calibrated prediction uncer- tainty in computational chemistry, The Journal of Chem- ical Physics156, 114109 (2022)
P. Pernot, The long road to calibrated prediction uncer- tainty in computational chemistry, The Journal of Chem- ical Physics156, 114109 (2022)
2022
-
[11]
Kellner and M
M. Kellner and M. Ceriotti, Uncertainty quantifica- tion by direct propagation of shallow ensembles, Mach. Learn.: Sci. Technol.5, 035006 (2024)
2024
-
[12]
F. Bigi, S. Chong, M. Ceriotti, and F. Grasselli, A pre- diction rigidity formalism for low-cost uncertainties in trained neural networks, Mach. Learn.: Sci. Technol.5, 045018 (2024)
2024
-
[13]
A. Zhu, S. Batzner, A. Musaelian, and B. Kozinsky, Fast uncertainty estimates in deep learning interatomic po- tentials, The Journal of Chemical Physics158, 164111 (2023)
2023
-
[14]
Perez, A
D. Perez, A. P. A. Subramanyam, I. Maliyov, and T. D. Swinburne, Uncertainty quantification for misspecified machine learned interatomic potentials, npj Computa- tional Materials11, 263 (2025)
2025
-
[15]
J. J. Mortensen, K. Kaasbjerg, S. L. Frederiksen, J. K. Nørskov, J. P. Sethna, and K. W. Jacobsen, Bayesian Error Estimation in Density-Functional Theory, Physical Review Letters95, 216401 (2005)
2005
-
[16]
Wellendorff, K
J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Pet- zold, D. D. Landis, J. K. Nørskov, T. Bligaard, and K. W. Jacobsen, Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation, Physical Review B85, 235149 (2012)
2012
-
[17]
Aldegunde, J
M. Aldegunde, J. R. Kermode, and N. Zabaras, De- velopment of an exchange–correlation functional with uncertainty quantification capabilities for density func- tional theory, Journal of Computational Physics311, 173 (2016)
2016
-
[18]
Wellendorff, K
J. Wellendorff, K. T. Lundgaard, K. W. Jacobsen, and T. Bligaard, mBEEF: An accurate semi-local Bayesian error estimation density functional, The Journal of Chemical Physics140, 144107 (2014)
2014
-
[19]
Hansen, J
T. Hansen, J. J. Mortensen, T. Bligaard, and K. W. Ja- cobsen, Uncertainty-aware electronic density-functional distributions, Physical Review B112, 075412 (2025)
2025
-
[20]
Pozdnyakov and M
S. Pozdnyakov and M. Ceriotti, Smooth, exact rotational symmetrization for deep learning on point clouds, inAd- vances in Neural Information Processing Systems, Vol. 36 (Curran Associates, Inc., 2023) pp. 79469–79501
2023
- [21]
-
[22]
M. Sch¨ afer, M. Kellner, J. K¨ astner, and M. Ceriotti, How to Train a Shallow Ensemble (2026), arXiv:2602.15747 [physics]
-
[23]
G. M. Torrie and J. P. Valleau, Nonphysical sampling dis- tributions in Monte Carlo free-energy estimation: Um- brella sampling, Journal of Computational Physics23, 187 (1977)
1977
-
[24]
M. Ceriotti, G. A. Brain, O. Riordan, and D. E. Manolopoulos, The inefficiency of re-weighted sampling and the curse of system size in high-order path integra- tion, Proceedings of the Royal Society A: Mathemati- cal, Physical and Engineering Sciences468, 2 (2012), arXiv:1107.1908
-
[25]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Physical Review Letters77, 3865 (1996)
1996
- [26]
-
[27]
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vy- drov, G. E. Scuseria, L. A. Constantin, X. Zhou, and K. Burke, Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Physical Review Letters 100, 136406 (2008)
2008
-
[28]
Mazitov, S
A. Mazitov, S. Chorna, G. Fraux, M. Bercx, G. Pizzi, S. De, and M. Ceriotti, Massive Atomic Diversity: A 12 compact universal dataset for atomistic machine learn- ing, Sci Data12, 1857 (2025)
2025
-
[29]
Barroso-Luque et al., Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
L. Barroso-Luque, M. Shuaibi, X. Fu, B. M. Wood, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick, and Z. W. Ulissi, Open Materials 2024 (OMat24) Inorganic Materi- als Dataset and Models (2024), arXiv:2410.12771 [cond- mat]
-
[30]
J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew, and J. Sun, Accurate and Numerically Efficient r2SCAN Meta-Generalized Gradient Approximation, The Journal of Physical Chemistry Letters11, 8208 (2020)
2020
-
[31]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A con- sistent and accurate ab initio parametrization of den- sity functional dispersion correction (DFT-D) for the 94 elements H-Pu, The Journal of Chemical Physics132, 154104 (2010)
2010
-
[32]
L. A. Curtiss, K. Raghavachari, P. C. Redfern, and J. A. Pople, Assessment of Gaussian-3 and density functional theories for a larger experimental test set, The Journal of Chemical Physics112, 7374 (2000)
2000
-
[33]
F. Tran, J. Stelzl, and P. Blaha, Rungs 1 to 4 of DFT Jacob’s ladder: Extensive test on the lattice constant, bulk modulus, and cohesive energy of solids, The Journal of Chemical Physics144, 204120 (2016)
2016
-
[34]
Bussi, D
G. Bussi, D. Donadio, and M. Parrinello, Canonical sam- pling through velocity rescaling, Journal of Chemical Physics126, 14101 (2007)
2007
-
[35]
Ceriotti, G
M. Ceriotti, G. Bussi, and M. Parrinello, Colored-noise thermostats ` a la Carte, Journal of Chemical Theory and Computation6, 1170 (2010)
2010
-
[36]
Bussi, T
G. Bussi, T. Zykova-Timan, and M. Parrinello, Isothermal-isobaric molecular dynamics using stochastic velocity rescaling, The Journal of Chemical Physics130, 074101 (2009)
2009
-
[37]
Ceriotti, J
M. Ceriotti, J. More, and D. E. Manolopoulos, I-PI: A Python interface for ab initio path integral molecular dy- namics simulations, Computer Physics Communications 185, 1019 (2014)
2014
-
[38]
Litman, V
Y. Litman, V. Kapil, Y. M. Y. Feldman, D. Tisi, T. Beguˇ si´ c, K. Fidanyan, G. Fraux, J. Higer, M. Kellner, T. E. Li, E. S. P´ os, E. Stocco, G. Trenins, B. Hirshberg, M. Rossi, and M. Ceriotti, I-PI 3.0: A flexible and effi- cient framework for advanced atomistic simulations, The Journal of Chemical Physics161, 062504 (2024)
2024
-
[39]
F. Bigi, J. W. Abbott, P. Loche, A. Mazitov, D. Tisi, M. F. Langer, A. Goscinski, P. Pegolo, S. Chong, R. Goswami, P. Febrer, S. Chorna, M. Kellner, M. Ceri- otti, and G. Fraux, Metatensor and metatomic : Founda- tional libraries for interoperable atomistic machine learn- ing, The Journal of Chemical Physics164, 064113 (2026)
2026
-
[40]
R. J. Gowers, M. Linke, J. Barnoud, T. J. E. Reddy, M. N. Melo, S. L. Seyler, J. Doma´ nski, D. L. Dotson, S. Buchoux, I. M. Kenney, and O. Beckstein, MDAnaly- sis: A Python Package for the Rapid Analysis of Molecu- lar Dynamics Simulations, SciPy 2016 10.25080/Majora- 629e541a-00e (2016)
-
[41]
Michaud-Agrawal, E
N. Michaud-Agrawal, E. J. Denning, T. B. Woolf, and O. Beckstein, MDAnalysis: A toolkit for the analysis of molecular dynamics simulations, Journal of Compu- tational Chemistry32, 2319 (2011)
2011
-
[42]
Petkov, Pair Distribution Functions Analysis, inChar- acterization of Materials(John Wiley & Sons, Ltd, 2012) pp
V. Petkov, Pair Distribution Functions Analysis, inChar- acterization of Materials(John Wiley & Sons, Ltd, 2012) pp. 1–14
2012
-
[43]
Tovey, A
S. Tovey, A. Narayanan Krishnamoorthy, G. Sivaraman, J. Guo, C. Benmore, A. Heuer, and C. Holm, DFT Ac- curate Interatomic Potential for Molten NaCl from Ma- chine Learning, The Journal of Physical Chemistry C 124, 25760 (2020)
2020
-
[44]
Ohno and K
H. Ohno and K. Furukawa, X-ray diffraction analysis of molten NaCl near its melting point, Journal of the Chem- ical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases77, 1981 (1981)
1981
-
[45]
Del Rio, Pascual, Rodriguez, Gonz´ alez, and Gonz´ alez, First principles determination of some static and dy- namic properties of the liquid 3d transition metals near melting, Condensed Matter Physics23, 23606 (2020)
2020
-
[46]
A. K. Soper, The radial distribution functions of water and ice from 220 to 673 K and at pressures up to 400 MPa, Chemical Physics258, 121 (2000)
2000
-
[47]
D. R. Lide, ed., Density of water at 1 atmosphere, inCRC Handbook of Chemistry and Physics(CRC Press, Boca Raton, FL, 2005) Chap. 6, pp. 6–5, internet version 2005 ed
2005
-
[48]
Bellissent, C
R. Bellissent, C. Bergman, R. Ceolin, and J. P. Gaspard, Structure of liquid As: A Peierls distortion in a liquid, Physical Review Letters59, 661 (1987)
1987
-
[49]
Klemm and H
W. Klemm and H. Niermann, Further Contributions to the Knowledge of Semimetals, Angewandte Chemie In- ternational Edition in English2, 523 (1963)
1963
-
[50]
M. C. Bellissent-Funel, P. Chieux, D. Levesque, and J. J. Weis, Structure factor and effective two-body potential for liquid gallium, Physical Review A39, 6310 (1989)
1989
-
[51]
Bergman, C
C. Bergman, C. Bichara, P. Chieux, and J. P. Gaspard, ON THE ATOMIC STRUCTURE OF LIQUID GaAs, Le Journal de Physique Colloques46, C8 (1985)
1985
-
[52]
V. M. Glazov, S. N. Chizhevskaya, and N. N. Glagoleva, Liquid Semiconductors(Plenum Press, 1969) p. 124
1969
-
[53]
Waseda, The structure of non-crystalline materials, Liquids and Amorphous Solids (1980)
Y. Waseda, The structure of non-crystalline materials, Liquids and Amorphous Solids (1980)
1980
-
[54]
H. Lou, X. Wang, Q. Cao, D. Zhang, J. Zhang, T. Hu, H.-k. Mao, and J.-Z. Jiang, Negative expansions of in- teratomic distances in metallic melts, Proceedings of the National Academy of Sciences110, 10068 (2013)
2013
-
[55]
Schmitz-Pranghe and R
N. Schmitz-Pranghe and R. Kohlhaas, Notizen: R¨ ontgenbeugungsuntersuchungen an Eisen, Kobalt und Nickel im fl¨ ussigen Zustand, Zeitschrift f¨ ur Natur- forschung A25, 1752 (1970)
1970
-
[56]
M. I. Mendelev, S. Han, D. J. Srolovitz, G. J. Ackland, D. Y. Sun, and M. Asta, Development of new inter- atomic potentials appropriate for crystalline and liquid iron, Philosophical Magazine83, 3977 (2003)
2003
-
[57]
Il’inskii, S
A. Il’inskii, S. Slyusarenko, O. Slukhovskii, I. Kaban, and W. Hoyer, Structure of liquid Fe–Al alloys, Materials Sci- ence and Engineering: A325, 98 (2002)
2002
-
[58]
Ntonti, S
E. Ntonti, S. Sotiriadou, M. J. Assael, M. L. Huber, B. Wilthan, and M. Watanabe, Reference Correlations for the Density and Thermal Conductivity, and Re- view of the Viscosity Measurements, of Liquid Titanium, Zirconium, Hafnium, Vanadium, Niobium, Tantalum, Chromium, Molybdenum, and Tungsten, International Journal of Thermophysics45, 18 (2024)
2024
-
[59]
Holland-Moritz, O
D. Holland-Moritz, O. Heinen, R. Bellissent, and T. Schenk, Short-range order of stable and undercooled liquid titanium, Materials Science and Engineering: A Proceedings of the 12th International Conference on Rapidly Quenched & Metastable Materials,449–451, 42 (2007). 13
2007
-
[60]
M. Mayo, E. Yahel, Y. Greenberg, and G. Makov, Short range order in liquid pnictides, Journal of Physics: Con- densed Matter25, 505102 (2013)
2013
-
[61]
Greenberg, E
Y. Greenberg, E. Yahel, E. N. Caspi, C. Benmore, B. Beuneu, M. P. Dariel, and G. Makov, Evidence for a temperature-driven structural transformation in liquid bismuth, Europhysics Letters86, 36004 (2009)
2009
-
[62]
Itami, S
T. Itami, S. Munejiri, T. Masaki, H. Aoki, Y. Ishii, T. Kamiyama, Y. Senda, F. Shimojo, and K. Hoshino, Structure of liquid Sn over a wide temperature range from neutron scattering experiments and first-principles molecular dynamics simulation: A comparison to liquid Pb, Physical Review B67, 064201 (2003)
2003
-
[63]
Greenberg, E
Y. Greenberg, E. Yahel, E. N. Caspi, B. Beuneu, M. P. Dariel, and G. Makov, On the relation between the mi- croscopic structure and the sound velocity anomaly in elemental melts of groups IV, V, and VI, The Journal of Chemical Physics133, 094506 (2010). 14 Supplementary Information S1. MLIP MODEL ARCHITECTURE AND TRAINING DETAILS In this work, an ensemble...
2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.