Recognition: unknown
Computational Design and Experimental Validation of Photoactive PARP1 Inhibitors
Pith reviewed 2026-05-07 17:48 UTC · model grok-4.3
The pith
Computational screening found a molecule that inhibits the PARP1 cancer target 15 times more strongly under green light.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
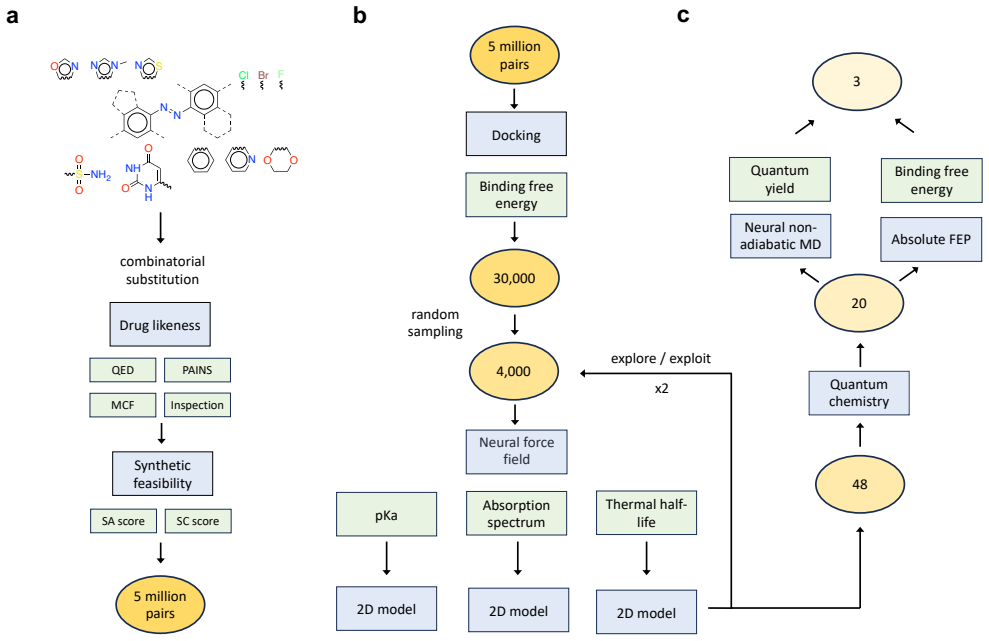
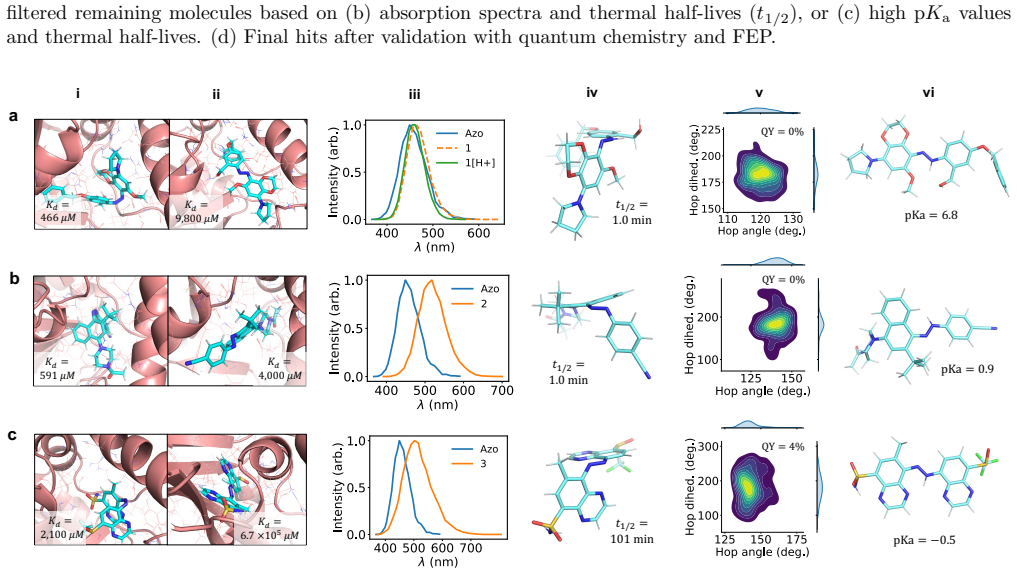
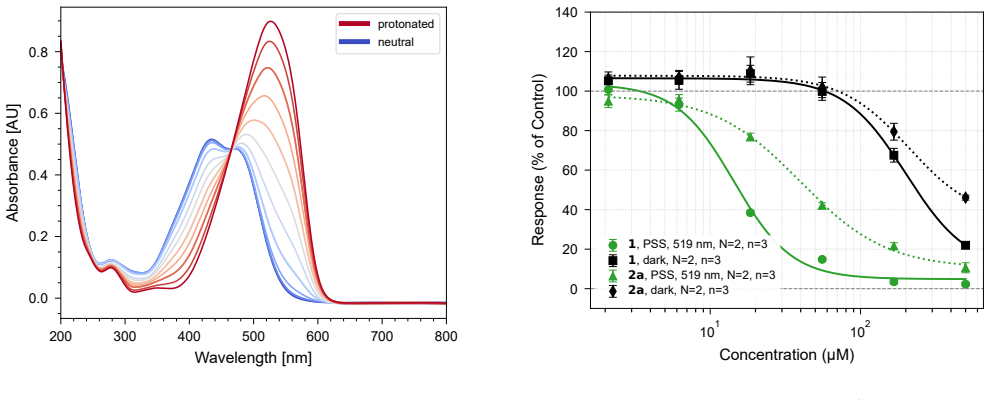
Using atomistic simulation and machine learning, we screened five million hypothetical photoactive ligands for PARP1 inhibition that depends on light-induced isomerization. Prioritizing candidates with favorable photophysical properties and differential binding, we synthesized ten molecules and found that compound 1 exhibits a 15-fold enhancement in PARP1 inhibition upon 519 nm irradiation (IC50 208.8 μM dark vs 14.4 μM light), validating the computation-guided strategy for identifying red-shifted PARP1 photoinhibitors while noting rapid thermal relaxation in aqueous media.
What carries the argument
The multi-stage computational workflow of protein-ligand docking for differential binding, ML force fields and quantum chemistry for spectra and half-lives, graph-based surrogates, excited-state dynamics for quantum yields, and free energy perturbation for binding refinement.
If this is right
- The validated workflow reduces the number of molecules that must be made and tested to find light-controlled inhibitors.
- Red-shifted absorption allows visible rather than ultraviolet light for activation, lowering risk of tissue damage.
- The approach extends to designing photoactive ligands for other protein targets in photopharmacology.
- Current limitations such as fast thermal relaxation in water point to needed improvements in isomer stability for biological use.
Where Pith is reading between the lines
- Light-activated PARP1 inhibitors could allow spatial control in tumors by shining light only on diseased tissue, sparing healthy cells.
- The observed rapid relaxation suggests testing the compounds in lipid environments or modifying the switching group for longer-lived states.
- The same screening pipeline could incorporate additional criteria like cell permeability or selectivity over related PARP family members.
- Success here lowers the barrier to applying computation-first design for other classes of photoresponsive drugs.
Load-bearing premise
The computational predictions of binding affinities, absorption spectra, thermal half-lives, and quantum yields accurately reflect the experimental behavior of the synthesized compounds.
What would settle it
An experiment in which the synthesized compounds show no measurable difference in PARP1 inhibition between dark and green-light conditions, or where measured absorption wavelengths and thermal half-lives deviate substantially from the predictions.
Figures


read the original abstract
Light-activated drugs are a promising way to treat localized diseases for which existing treatments have severe side effects. However, their development is complicated by the set of photophysical and biological properties that must be simultaneously optimized. Here we used computational techniques to find a set of promising candidates for the photoactive inhibition of the poly(ADP-ribose) polymerase 1 (PARP1) cancer target. Using our recently developed methods based on atomistic simulation and machine learning (ML), we screened a set of 5 million hypothetical photoactive ligands. Our workflow used protein-ligand docking to identify candidates with differential PARP1 binding under light and dark conditions; ML force fields and quantum chemistry calculations to predict p$K_\mathrm{a}$, absorption spectra, and thermal half-lives; graph-based surrogate models to screen additional compounds; excited-state nonadiabatic dynamics with ML force fields to estimate quantum yields; and free energy perturbation (FEP) to refine binding predictions. From these predictions, we prioritized a small set of synthetically feasible candidates expected to have red-shifted absorption spectra, thermal half-lives on the order of seconds to minutes, and isomer-dependent PARP1 binding under visible-light control. We synthesized 10 candidates and experimentally characterized their photobehavior and PARP1 inhibition constants. Among the validated compounds, \textbf{1} showed a 15-fold increase in inhibition of PARP1 upon green-light irradiation at 519 nm (208.8 $\pm$ 28.3 $\mu$M vs 14.4 $\pm$ 1.9 $\mu$M). These results validate the computation-guided screening strategy for identifying red-shifted PARP1 photoinhibitors, while also underscoring current limitations such as rapid thermal relaxation in aqueous media.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper describes a multi-stage computational pipeline (docking for differential binding, ML force fields + QC for pKa/spectra/half-lives, graph surrogates, nonadiabatic dynamics for quantum yields, and FEP) to screen 5 million hypothetical photoactive ligands for red-shifted PARP1 inhibitors. Ten synthetically feasible candidates were prioritized, synthesized, and tested; compound 1 exhibited a 15-fold light-dependent increase in PARP1 inhibition at 519 nm (208.8 ± 28.3 μM dark vs. 14.4 ± 1.9 μM irradiated), while the abstract notes limitations including rapid thermal relaxation in aqueous media. The authors conclude that the results validate the computation-guided strategy.
Significance. The experimental synthesis and testing of ten compounds, yielding a clear functional photoinhibitor with quantified 15-fold activity change, provides direct and independent support for identifying light-activated PARP1 inhibitors suitable for localized cancer treatment. This is a concrete advance. However, the significance for validating computational design methods is limited because the manuscript does not report quantitative agreement between predicted and measured photophysical properties, leaving open whether the pipeline enriched for success through accurate physics or by chance.
major comments (2)
- [Abstract] Abstract: the claim that 'These results validate the computation-guided screening strategy' is not supported by evidence in the manuscript. No table or section compares the computationally predicted absorption spectra (λ_max), thermal half-lives (t1/2), or quantum yields (Φ) against the experimentally measured values for compound 1 or the other nine synthesized candidates. Without this comparison, it cannot be determined whether the multi-stage pipeline (docking, ML force fields, nonadiabatic dynamics) correctly captured the relevant photophysics or whether the observed 15-fold effect occurred despite inaccurate predictions.
- [Results] Results (experimental characterization of photobehavior): the inhibition constants for compound 1 are reported with standard deviations and a clear 15-fold change, but the photophysical characterization (e.g., measured λ_max at 519 nm, thermal relaxation rates in aqueous media) is presented without direct side-by-side comparison to the prior computational predictions used for prioritization. This omission is load-bearing for the central claim that the workflow successfully identified functional red-shifted photoinhibitors.
minor comments (2)
- [Abstract and Methods] The abstract and methods could more explicitly state which prior author publications the ML force fields and graph surrogates are reused from, to allow readers to assess the novelty of the current workflow.
- [Figures] Figure captions for the inhibition data should include the exact irradiation wavelength, intensity, and duration to ensure reproducibility of the 519 nm experiment.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and for recognizing the significance of the experimental validation of compound 1 as a photoactive PARP1 inhibitor. We address each major comment below and commit to revisions that will provide the requested comparisons to better support our claims about the computational strategy.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that 'These results validate the computation-guided screening strategy' is not supported by evidence in the manuscript. No table or section compares the computationally predicted absorption spectra (λ_max), thermal half-lives (t1/2), or quantum yields (Φ) against the experimentally measured values for compound 1 or the other nine synthesized candidates. Without this comparison, it cannot be determined whether the multi-stage pipeline (docking, ML force fields, nonadiabatic dynamics) correctly captured the relevant photophysics or whether the observed 15-fold effect occurred despite inaccurate predictions.
Authors: We agree that including quantitative comparisons between predicted and experimental photophysical properties would provide stronger evidence for the accuracy of the computational pipeline. The current manuscript focuses on the successful experimental outcome following computational prioritization, but does not present direct side-by-side data. In the revised version, we will add a dedicated section or table comparing the computed λ_max, t1/2, and Φ (where experimental data are available) with the measured values for the ten synthesized compounds. We will also moderate the abstract claim to reflect that the results support the utility of the strategy while highlighting the need for further validation of individual predictions. revision: yes
-
Referee: [Results] Results (experimental characterization of photobehavior): the inhibition constants for compound 1 are reported with standard deviations and a clear 15-fold change, but the photophysical characterization (e.g., measured λ_max at 519 nm, thermal relaxation rates in aqueous media) is presented without direct side-by-side comparison to the prior computational predictions used for prioritization. This omission is load-bearing for the central claim that the workflow successfully identified functional red-shifted photoinhibitors.
Authors: We acknowledge this point and will revise the Results section to include direct comparisons of the experimental photophysical data (such as the observed absorption maximum at 519 nm and thermal relaxation rates) with the corresponding computational predictions used in the screening workflow. This will allow readers to evaluate the performance of the multi-stage pipeline more rigorously. Note that some properties like quantum yields may not have been experimentally quantified, which we will explicitly state as a limitation. revision: yes
- We did not measure quantum yields experimentally for the synthesized compounds, so a full comparison for Φ cannot be provided; this will be noted in the revisions.
Circularity Check
Minor self-citation of prior methods; central experimental validation remains independent
full rationale
The paper applies previously published computational workflows (ML force fields, nonadiabatic dynamics, FEP) to screen candidates and then reports direct experimental measurements of inhibition constants and photobehavior for the synthesized compounds. The load-bearing claim is the 15-fold light-dependent inhibition observed for compound 1, which is measured data and does not reduce to any fitted parameter or self-cited equation by construction. Self-citation of 'our recently developed methods' occurs but is not load-bearing for the final result, as the experimental outcome stands on its own. No self-definitional, fitted-input-as-prediction, or ansatz-smuggling steps are present in the derivation chain.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
(1) Lerch, M. M.; Hansen, M. J.; van Dam, G. M.; Szymanski, W.; Feringa, B. L. Emerging targets in photophar- macology.Angewandte Chemie International Edition2016,55, 10978–10999. (2) Broichhagen, J.; Frank, J. A.; Trauner, D. A roadmap to success in photopharmacology.Accounts of chemical research2015,48, 1947–1960. (3) Szymanski, W.; Ourailidou, M. E.; V...
-
[2]
(17) Axelrod, S.; Shakhnovich, E.; G´ omez-Bombarelli, R. Thermal half-lives of azobenzene derivatives: Virtual screening based on intersystem crossing using a machine learning potential.ACS Central Science2023,9, 166–176. (18) Dong, M.; Babalhavaeji, A.; Hansen, M.; Kalman, L.; Woolley, G. Red, far-red, and near infrared photo- switches based on azonium ...
-
[3]
Recent developments in the general atomic and molecular electronic structure system,
(29) RDKit: Open-source cheminformatics.http://www.rdkit.org. (30) Garc´ ıa-Orteg´ on, M.; Simm, G. N.; Tripp, A. J.; Hern´ andez-Lobato, J. M.; Bender, A.; Bacallado, S. DOCK- STRING: easy molecular docking yields better benchmarks for ligand design.Journal of chemical information and modeling2022,62, 3486–3502. (31) YANK.https://github.com/choderalab/ya...
-
[4]
R.; Fargo, J
(51) Talaty, E. R.; Fargo, J. C. Thermal cis–trans-isomerization of substituted azobenzenes: a correction of the literature.Chemical Communications (London)1967, 65–66. 22 (52) Lee, S.; Filatov, M.; Lee, S.; Choi, C. H. Eliminating spin-contamination of spin-flip time dependent density functional theory within linear response formalism by the use of zerot...
1967
-
[5]
(75) Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms.Journal of Physics: Condensed Matter2017,29, 273002. (76) PDBFixer.https://github.com/openmm/pdbfixer, Accessed: 2023-04-19. (77) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.; Klein, M. L. Comparison of simple potential functions for sim...
-
[6]
N.; Kaiser, L.; Polosukhin, I
(100) Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A. N.; Kaiser, L.; Polosukhin, I. Attention is all you need. Advances in neural information processing systems. 2017; pp 5998–6008. (101) Gilmer, J.; Schoenholz, S. S.; Riley, P. F.; Vinyals, O.; Dahl, G. E. Neural message passing for quantum chemistry. International conference o...
2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.