Recognition: unknown
Mixture of Experts Framework in Machine Learning Interatomic Potentials for Atomistic Simulations
Pith reviewed 2026-05-07 13:44 UTC · model grok-4.3
The pith
Co-trained mixture-of-experts interatomic potentials partition complex regions from bulk to run atomistic simulations more than twice as fast while conserving energy exactly.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By spatially partitioning the domain and co-training independent E(3)-equivariant Allegro models with agreement constraints on bulk environments, the mixture-of-experts models maintain exact energy conservation, align their bulk mechanical responses including equation of state and bulk modulus, and match the accuracy of a uniform high-fidelity simulation at more than twice the speed on a realistic Pt+CO catalytic system.
What carries the argument
The co-training loss that penalizes per-atom energy and force discrepancies between models evaluated on shared bulk environments, inside a static spatial partition of the simulation domain.
If this is right
- Large-scale atomistic simulations become feasible at high accuracy without a proportional increase in cost.
- Exact energy conservation supports stable dynamics over long timescales.
- High-fidelity training data can be limited to the complex region while the bulk region uses a cheaper model.
- Mechanical properties such as bulk modulus remain consistent across the entire domain.
Where Pith is reading between the lines
- The same co-training idea could support adaptive rather than static partitioning that activates the high-fidelity model only near defects or reactions as they appear.
- Hybrid simulations that combine quantum-mechanical calculations in the complex region with the lower-capacity model in bulk become more practical if the agreement constraint generalizes.
- Systems containing phase boundaries or extended defects would test whether the static-partition assumption continues to hold without additional interface corrections.
Load-bearing premise
A static spatial partition combined with bulk-environment agreement constraints is sufficient to eliminate interface-induced stress fields and instability across arbitrary material systems and reactive conditions.
What would settle it
A long molecular-dynamics trajectory on a system whose reactive zones move or whose interface chemistry deviates from the bulk training environments produces growing artificial stress or energy drift.
Figures
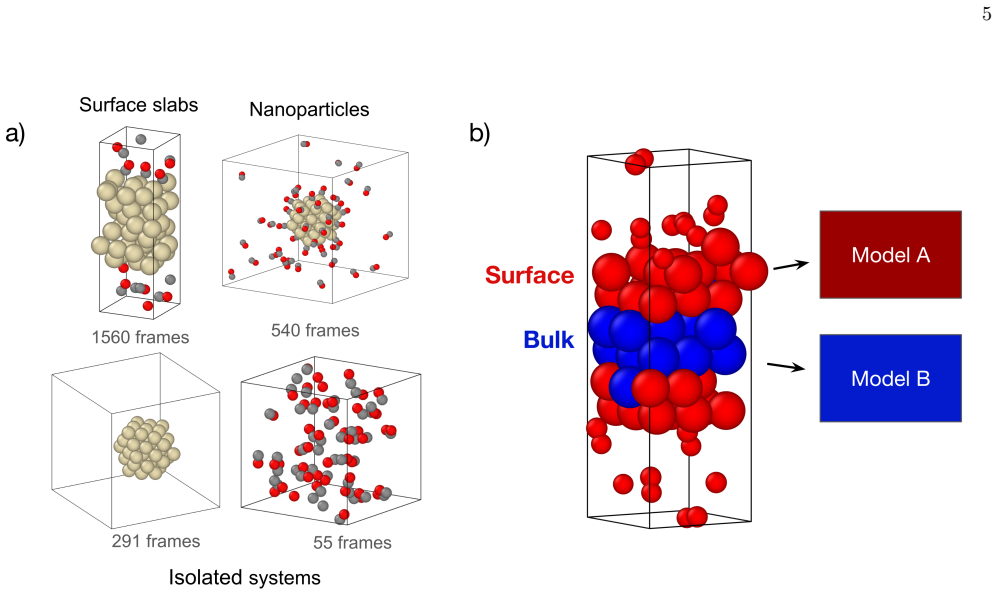


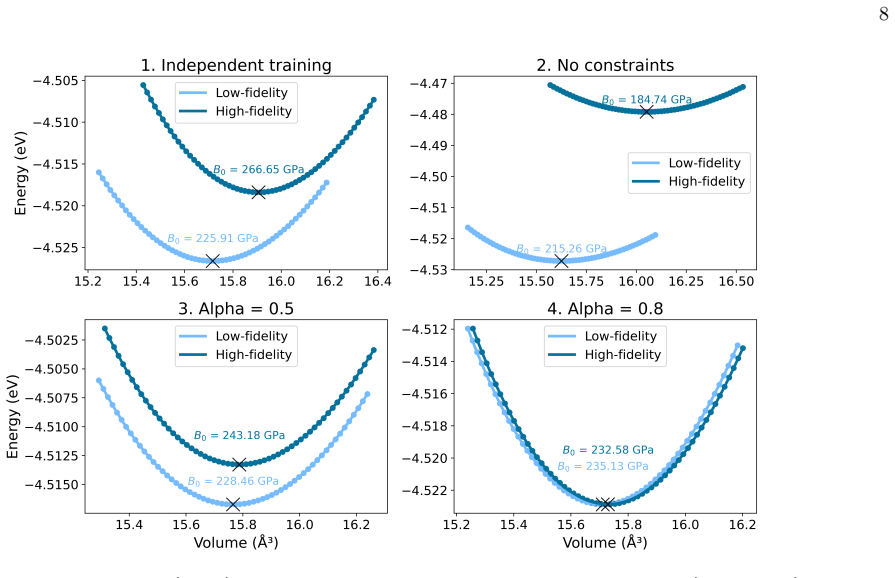
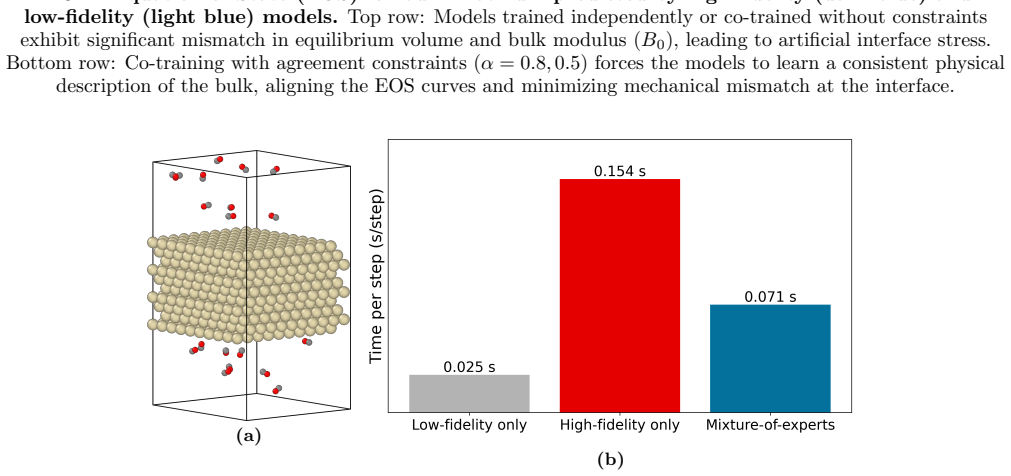
read the original abstract
First-principles atomistic simulations are essential for understanding complex material phenomena but are fundamentally limited by their computational cost. While Machine Learning Interatomic Potentials (MLIPs) have drastically improved cost for a given accuracy, their inference cost remains a bottleneck for massive systems or long timescales. To address this, we introduce a multifidelity "Mixture-of-Experts" framework based on the E(3)-equivariant Allegro architecture. Our method spatially partitions the simulation domain into a chemically complex region (e.g., reactive interfaces) and a simple region (e.g., bulk lattice), assigning models of varying capacity to each. Among the challenges in such static domain decomposition, the mechanical mismatch between models at the interface is particularly critical, as it can generate artificial stress fields and instability. We address this challenge with a co-training strategy in which the loss function includes agreement constraints -- penalties on per-atom energy and force discrepancies between models evaluated on shared bulk environments -- forcing the independent models to learn a consistent physical description of the bulk material. We validate this approach on a realistic Pt+CO catalytic system, demonstrating that the co-trained models maintain exact energy conservation, align their bulk mechanical response (e.g., equation of state and bulk modulus), and achieve predictive accuracy comparable to a full high-fidelity simulation at more than twice the computational speed.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a multifidelity Mixture-of-Experts framework for ML interatomic potentials based on the E(3)-equivariant Allegro architecture. The simulation domain is statically partitioned into a chemically complex region (e.g., reactive interfaces) handled by a high-capacity model and a simple bulk region handled by a lower-capacity model. A co-training strategy augments the loss with agreement penalties on per-atom energies and forces evaluated on shared bulk environments to enforce mechanical consistency between the independent models. Validation on a Pt+CO catalytic system is claimed to show exact energy conservation, alignment of bulk mechanical properties (equation of state and bulk modulus), predictive accuracy comparable to a full high-fidelity simulation, and more than twice the computational speed.
Significance. If the central claims hold, the approach would enable substantially larger-scale or longer-timescale atomistic simulations of systems with localized reactive zones by reducing inference cost without compromising stability or accuracy. The co-training mechanism for interface consistency is a targeted solution to a known difficulty in domain-decomposed MLIPs and, if shown to be robust, would be a useful addition to the toolkit for catalysis and materials modeling.
major comments (3)
- [Abstract and validation section] Abstract and validation section: the claims of 'predictive accuracy comparable to a full high-fidelity simulation' and 'more than twice the computational speed' are stated without any quantitative error metrics (e.g., MAE or RMSE on energies/forces), baseline comparisons to single-model or other multifidelity approaches, training-set sizes, or hyperparameter details, so it is not possible to verify that the reported results support the performance assertions.
- [Co-training strategy description] Co-training strategy description: agreement constraints are imposed only on per-atom energies and forces from shared bulk environments, yet atoms whose local neighborhoods straddle the static partition boundary possess descriptors that differ from the pure-bulk training data; the manuscript provides no proof or numerical test that the resulting effective potential and its derivatives remain continuous across this boundary.
- [Validation results on Pt+CO] Validation results on Pt+CO: while bulk equation-of-state and modulus alignment are reported, no interface-stress diagnostics, force-jump statistics, or stability metrics for atoms at or crossing the partition boundary are supplied, leaving untested whether the co-training suffices to suppress artificial stress fields under the reactive conditions the method is intended to address.
minor comments (2)
- [Methods] The precise mathematical form of the agreement penalty term in the co-training loss should be written explicitly (e.g., as an additional term in the total loss function) to allow readers to reproduce the weighting between data fidelity and model-consistency objectives.
- [Methods] A schematic diagram illustrating the spatial partition, the assignment of experts, and the locations at which agreement constraints are evaluated would improve clarity of the domain-decomposition procedure.
Simulated Author's Rebuttal
We thank the referee for their insightful and constructive comments on our manuscript. We have addressed each of the major comments point by point below, making revisions to the manuscript where necessary to strengthen the presentation of our results.
read point-by-point responses
-
Referee: [Abstract and validation section] Abstract and validation section: the claims of 'predictive accuracy comparable to a full high-fidelity simulation' and 'more than twice the computational speed' are stated without any quantitative error metrics (e.g., MAE or RMSE on energies/forces), baseline comparisons to single-model or other multifidelity approaches, training-set sizes, or hyperparameter details, so it is not possible to verify that the reported results support the performance assertions.
Authors: We agree that the abstract and validation section would benefit from more explicit quantitative metrics to support the performance claims. In the revised manuscript, we have added specific values for the mean absolute errors (MAE) and root mean square errors (RMSE) on energies and forces for the mixture-of-experts model compared to the full high-fidelity model. We also include details on the training set sizes used for each model and key hyperparameters. Baseline comparisons are provided, confirming that the accuracy is comparable (within acceptable thresholds for the application) while achieving the reported computational speedup. revision: yes
-
Referee: [Co-training strategy description] Co-training strategy description: agreement constraints are imposed only on per-atom energies and forces from shared bulk environments, yet atoms whose local neighborhoods straddle the static partition boundary possess descriptors that differ from the pure-bulk training data; the manuscript provides no proof or numerical test that the resulting effective potential and its derivatives remain continuous across this boundary.
Authors: The referee correctly identifies a potential issue with continuity at the partition boundary. Although the co-training focuses on bulk environments, the design assigns atoms with straddling neighborhoods to the high-fidelity model. We have performed additional numerical tests, now included in the revised manuscript, that sample atomic configurations crossing the boundary and verify that the effective potential and its derivatives (forces) exhibit continuity, with discrepancies below the level of thermal fluctuations. This supports the mechanical consistency of the approach. revision: yes
-
Referee: [Validation results on Pt+CO] Validation results on Pt+CO: while bulk equation-of-state and modulus alignment are reported, no interface-stress diagnostics, force-jump statistics, or stability metrics for atoms at or crossing the partition boundary are supplied, leaving untested whether the co-training suffices to suppress artificial stress fields under the reactive conditions the method is intended to address.
Authors: We acknowledge that the original validation focused primarily on bulk properties and overall accuracy. To address this, the revised manuscript now includes interface-specific diagnostics: stress tensor analysis near the boundary, statistics on force jumps for atoms crossing the partition, and long-term stability metrics from molecular dynamics simulations under reactive conditions. These additions demonstrate that artificial stress fields are effectively suppressed, with no significant instabilities observed. revision: yes
Circularity Check
No significant circularity; empirical validation is independent
full rationale
The paper defines an explicit MoE architecture with static spatial partitioning and a co-training loss that includes agreement penalties on per-atom energies/forces evaluated on shared bulk environments. These elements are design choices in the method, not tautological. The central claims (energy conservation, bulk mechanical alignment, and speed/accuracy on Pt+CO) are supported by direct comparison to a full high-fidelity simulation on an external validation system rather than reducing by construction to fitted inputs or self-citations. No load-bearing step invokes a uniqueness theorem, renames a known result, or smuggles an ansatz via prior self-work. The derivation chain is self-contained with external falsifiability.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The E(3)-equivariant Allegro architecture is an appropriate base model for both high- and low-fidelity experts.
Reference graph
Works this paper leans on
- [1]
-
[2]
E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nature communica- tions, 13(1):2453, 2022
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E Smidt, and Boris Kozinsky. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials.Nature communica- tions, 13(1):2453, 2022
2022
-
[3]
Mace: Higher order equiv- ariant message passing neural networks for fast and ac- curate force fields
Ilyes Batatia, David P Kovacs, Gregor Simm, Christoph Ortner, and Gabor Csanyi. Mace: Higher order equiv- ariant message passing neural networks for fast and ac- curate force fields. In S. Koyejo, S. Mohamed, A. Agar- wal, D. Belgrave, K. Cho, and A. Oh, editors,Advances in Neural Information Processing Systems, volume 35, pages 11423–11436. Curran Associ...
2022
-
[4]
Graph atomic cluster expansion for semilocal interactions beyond equivariant message passing.Physical Review X, 14(2):021036, 2024
Anton Bochkarev, Yury Lysogorskiy, and Ralf Drautz. Graph atomic cluster expansion for semilocal interactions beyond equivariant message passing.Physical Review X, 14(2):021036, 2024
2024
-
[5]
Warshel and M
A. Warshel and M. Levitt. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme.Journal of Molecular Biology, 103(2):227–249, 1976
1976
-
[6]
Hybrid atomistic simulation methods for materials sys- tems.Reports on Progress in Physics, 72(2):026501, 2009
Noam Bernstein, James R Kermode, and Gabor Csanyi. Hybrid atomistic simulation methods for materials sys- tems.Reports on Progress in Physics, 72(2):026501, 2009
2009
-
[7]
Mixture of diverse size experts
Manxi Sun, Wei Liu, Jian Luan, Pengzhi Gao, and Bin Wang. Mixture of diverse size experts. In Franck Dernon- court, Daniel Preot ¸iuc-Pietro, and Anastasia Shimorina, editors,Proceedings of the 2024 Conference on Empir- ical Methods in Natural Language Processing: Industry Track, pages 1608–1621, Miami, Florida, US, November
2024
-
[8]
Association for Computational Linguistics
-
[9]
Hmoe: Heterogeneous mixture of 10 experts for language modeling
An Wang, Xingwu Sun, Ruobing Xie, Shuaipeng Li, Jiaqi Zhu, Zhen Yang, Pinxue Zhao, Weidong Han, Zhanhui Kang, Di Wang, et al. Hmoe: Heterogeneous mixture of 10 experts for language modeling. InProceedings of the 2025 Conference on Empirical Methods in Natural Language Processing, pages 21954–21968, 2025
2025
-
[10]
Swinburne, and James R
Fraser Birks, Matthew Nutter, Thomas D. Swinburne, and James R. Kermode. Efficient and accurate spa- tial mixing of machine learned interatomic potentials for materials science.npj Computational Materials, 12(1), February 2026
2026
-
[11]
Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon.npj Computational Materials, 7(1):97, June 2021
Yury Lysogorskiy, Cas van der Oord, Anton Bochkarev, Sarath Menon, Matteo Rinaldi, Thomas Hammer- schmidt, Matous Mrovec, Aidan Thompson, G´ abor Cs´ anyi, Christoph Ortner, and Ralf Drautz. Performant implementation of the atomic cluster expansion (PACE) and application to copper and silicon.npj Computational Materials, 7(1):97, June 2021
2021
-
[12]
Xie, Matthias Rupp, and Richard G
Stephen R. Xie, Matthias Rupp, and Richard G. Hennig. Ultra-fast interpretable machine-learning potentials.npj Computational Materials, 9(1):162, September 2023
2023
-
[13]
The embedded-atom method: a review of theory and applications.Materials Science Reports, 9(7-8):251–310, 1993
Murray S Daw, Stephen M Foiles, and Michael I Baskes. The embedded-atom method: a review of theory and applications.Materials Science Reports, 9(7-8):251–310, 1993
1993
-
[14]
Adaptive-precision potentials for large-scale atomistic simulations.The Journal of Chemical Physics, 162(11), 2025
David Immel, Ralf Drautz, and Godehard Sutmann. Adaptive-precision potentials for large-scale atomistic simulations.The Journal of Chemical Physics, 162(11), 2025
2025
-
[15]
Conservative adaptive-precision interatomic potentials, December 2025
David Immel, Ralf Drautz, and Godehard Sutmann. Conservative adaptive-precision interatomic potentials, December 2025. arXiv:2512.07693 [physics]
-
[16]
Kitchin, Daniel S
Brandon M Wood, Misko Dzamba, Xiang Fu, Meng Gao, Muhammed Shuaibi, Luis Barroso-Luque, Kareem Abdelmaqsoud, Vahe Gharakhanyan, John R. Kitchin, Daniel S. Levine, Kyle Michel, Anuroop Sriram, Taco Cohen, Abhishek Das, Sushree Jagriti Sahoo, Ammar Rizvi, Zachary Ward Ulissi, and C. Lawrence Zitnick. UMA: A family of universal models for atoms. InThe Thirty...
2026
-
[17]
Yuzhi Liu, Duo Zhang, Anyang Peng, Weinan E, Lin- feng Zhang, and Han Wang. Scaling machine learning interatomic potentials with mixtures of experts.arXiv preprint arXiv:2603.07977, 2026
-
[18]
Zhang et al., A Graph Neural Network for the Era of Large Atomistic Models
Duo Zhang, Anyang Peng, Chengqian Cai, Wenshuo Li, Yu Zhou, Jinzhe Zeng, Mingyu Guo, Chengjian Zhang, Bowen Li, Hangrui Jiang, et al. Graph neural net- work model for the era of large atomistic models.arXiv preprint arXiv:2506.01686, 2025
-
[19]
Learning local equivariant representa- tions for large-scale atomistic dynamics.Nature Com- munications, 14(1):579, 2023
Albert Musaelian, Simon Batzner, Anders Johansson, Lixin Sun, Cameron J Owen, Mordechai Kornbluth, and Boris Kozinsky. Learning local equivariant representa- tions for large-scale atomistic dynamics.Nature Com- munications, 14(1):579, 2023
2023
-
[20]
Descoteaux, Mit Kotak, Gabriel de Miranda Nascimento, Se´ an R
Chuin Wei Tan, Marc L. Descoteaux, Mit Kotak, Gabriel de Miranda Nascimento, Se´ an R. Kavanagh, Laura Zichi, Menghang Wang, Aadit Saluja, Yizhong R. Hu, Tess Smidt, Anders Johansson, William C. Witt, Boris Kozin- sky, and Albert Musaelian. High-performance training and inference for deep equivariant interatomic potentials. Digital Discovery, 2026. Advanc...
2026
-
[21]
Surface rough- ening in nanoparticle catalysts.arXiv preprint arXiv:2407.13643, 2024
Cameron J Owen, Nicholas Marcella, Christopher R O’Connor, Taek-Seung Kim, Ryuichi Shimogawa, Clare Yijia Xie, Ralph G Nuzzo, Anatoly I Frenkel, Christian Reece, and Boris Kozinsky. Surface rough- ening in nanoparticle catalysts.arXiv preprint arXiv:2407.13643, 2024
-
[22]
On-the-fly active learning of interpretable bayesian force fields for atomistic rare events.npj Com- putational Materials, 6(1):20, 2020
Jonathan Vandermause, Steven B Torrisi, Simon Batzner, Yu Xie, Lixin Sun, Alexie M Kolpak, and Boris Kozinsky. On-the-fly active learning of interpretable bayesian force fields for atomistic rare events.npj Com- putational Materials, 6(1):20, 2020
2020
-
[23]
Active learning of reactive bayesian force fields applied to heterogeneous catalysis dynamics of h/pt.Nature Communications, 13(1):5183, 2022
Jonathan Vandermause, Yu Xie, Jin Soo Lim, Cameron J Owen, and Boris Kozinsky. Active learning of reactive bayesian force fields applied to heterogeneous catalysis dynamics of h/pt.Nature Communications, 13(1):5183, 2022
2022
-
[24]
Efficiency of ab- initio total energy calculations for metals and semicon- ductors using a plane-wave basis set.Computational ma- terials science, 6(1):15–50, 1996
Georg Kresse and J¨ urgen Furthm¨ uller. Efficiency of ab- initio total energy calculations for metals and semicon- ductors using a plane-wave basis set.Computational ma- terials science, 6(1):15–50, 1996
1996
-
[25]
Chemical accuracy for the van der waals density functional.Journal of Physics: Condensed Matter, 22(2):022201, dec 2009
Jiˇ r´ ı Klimeˇ s, David R Bowler, and Angelos Michaelides. Chemical accuracy for the van der waals density functional.Journal of Physics: Condensed Matter, 22(2):022201, dec 2009
2009
-
[26]
The atomic simulation environ- ment—a python library for working with atoms.Journal of Physics: Condensed Matter, 29(27):273002, 2017
Ask Hjorth Larsen, Jens Jørgen Mortensen, Jakob Blomqvist, Ivano E Castelli, Rune Christensen, Marcin Du lak, Jesper Friis, Michael N Groves, Bjørk Hammer, Cory Hargus, et al. The atomic simulation environ- ment—a python library for working with atoms.Journal of Physics: Condensed Matter, 29(27):273002, 2017
2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.