Recognition: unknown
Stepping up enhanced rate calculations with EATR-flooding
Pith reviewed 2026-05-07 10:28 UTC · model grok-4.3
The pith
Stepping up the strength of a biasing potential across separate simulation sets yields accurate rate constants for slow biomolecular processes without requiring time-dependent variation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
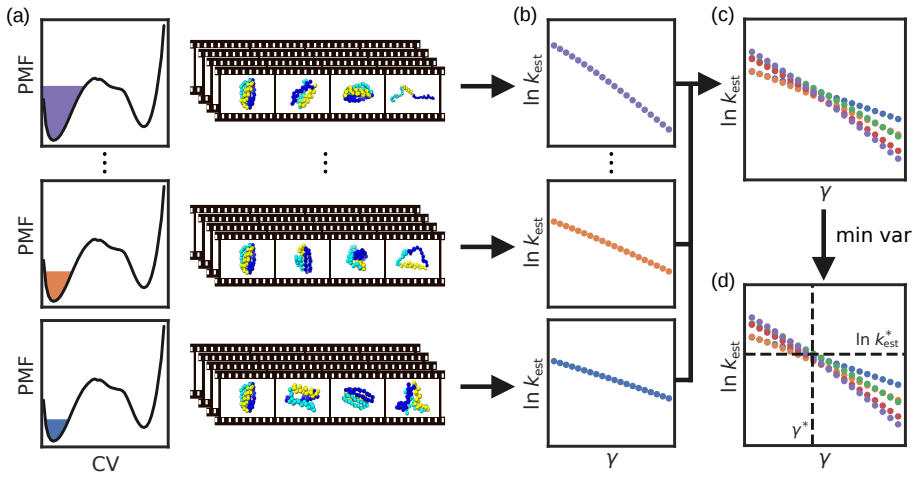
EATR-flooding replaces the time-dependent bias requirement of the original EATR method with a series of independent simulations performed at stepped-up values of the biasing potential. This change allows the same gamma-based correction for collective-variable quality to be applied, resulting in accurate rate estimates for quasi-static biasing schemes such as flooding and OPES while producing only one gamma value per choice of collective variables.
What carries the argument
EATR-flooding, which uses multiple simulation sets with increasing bias strengths to capture the same biasing-efficiency information previously obtained from continuous time variation, thereby keeping the gamma correction valid.
If this is right
- Rate calculations become feasible for enhanced-sampling schemes that cannot vary the bias continuously over time.
- Only a single gamma parameter needs to be determined for any given set of collective variables.
- An internal consistency check for over-biasing becomes available directly from the stepped simulation data.
- The approach extends to any collective-variable biasing method, not only OPES or flooding.
- Efficiency remains comparable to standard EATR on both coarse-grained and atomistic models.
Where Pith is reading between the lines
- Researchers limited to static-bias tools can now use the gamma correction without implementing time-dependent protocols.
- The single-gamma output may simplify systematic testing and selection of collective variables by removing dependence on bias schedule.
- The stepped-bias structure could be combined with other rate estimators that already operate on fixed-bias trajectories.
Load-bearing premise
Stepping up bias strength in separate runs captures equivalent information about how efficiently the bias modifies the observed rate as a continuously varying bias does.
What would settle it
Apply both standard EATR and EATR-flooding to the same coarse-grained protein folding system with known reference rates and check whether the two methods produce statistically indistinguishable results after gamma correction.
Figures





read the original abstract
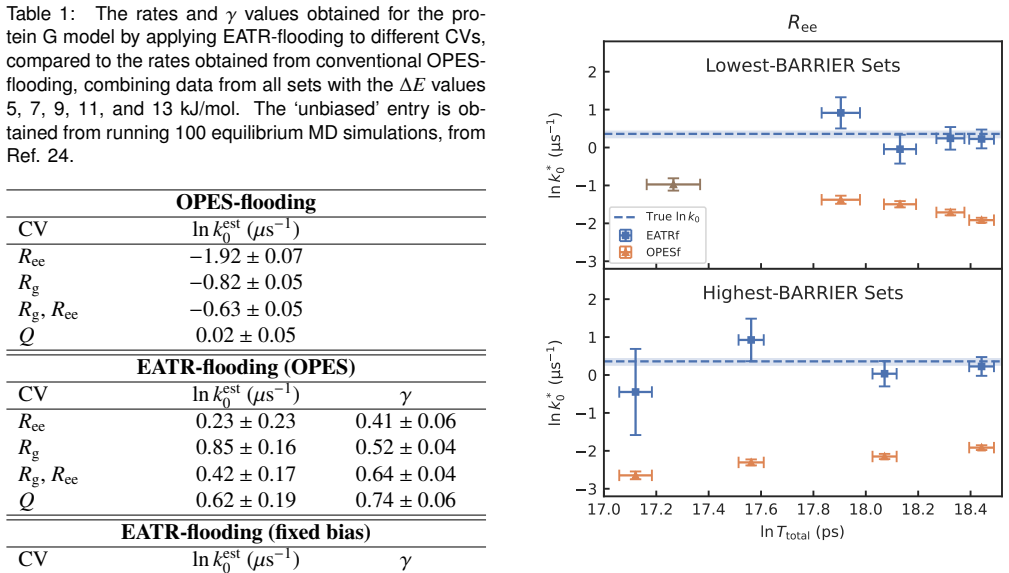
Several recent methods have shown that it is possible to compute rate constants of very slow biomolecular processes using simulations where a time-dependent bias is added along one or several collective variables (CVs). We previously reported the exponential average time-dependent rate (EATR) method, which can improve upon these approaches by accounting for how efficiently the external biasing potential modifies the observed rate using a learned CV-quality factor $\gamma$. This results in more accurate rate estimates using the same data when biasing a suboptimal coordinate. However, as formulated EATR depended on the biasing potential varying over time to properly determine the biasing efficiency, which limits the method's applicability to quasi-static biasing schemes such as ``flooding'' or on-the-fly probability enhanced sampling (OPES). Here, we present the EATR-flooding approach, which generalizes our method by replacing the need for a time dependent bias by instead varying (stepping up) the strength of the biasing potential across multiple sets of simulations. We implement this approach as an open-source Python library, and demonstrate that this approach is accurate without substantial loss of efficiency compared to standard EATR for a coarse-grained protein system, and also show good performance on a fully atomistic cavity-ligand model. Two additional appealing features of EATR-flooding are an internal check for over-biasing and the fact that only a single $\gamma$ parameter is predicted for a given choice of CVs, as compared to our earlier results where $\gamma$ empirically depended on biasing rate. Finally, we believe EATR-flooding applies not only to OPES simulations but more generally to CV biasing enhanced sampling approaches, making it broadly useful.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces EATR-flooding as a generalization of the prior EATR method for computing rate constants of slow biomolecular processes. It replaces the requirement for a continuously time-dependent bias with multiple independent simulation sets that step up the strength of a fixed biasing potential along collective variables. The approach is implemented as an open-source Python library and is claimed to yield accurate rates without substantial efficiency loss relative to standard EATR, as shown on a coarse-grained protein system and an atomistic cavity-ligand model. Additional features include an internal over-biasing check and a single gamma parameter per choice of CVs.
Significance. If the central transfer of the gamma correction holds, the work broadens the applicability of enhanced-sampling rate calculations to quasi-static protocols such as flooding and OPES. The open-source library and explicit demonstrations on both coarse-grained and fully atomistic models are concrete strengths that support reproducibility. The single-gamma property removes an earlier empirical dependence on biasing rate and could make the method more practical for suboptimal CVs.
major comments (1)
- [EATR-flooding generalization (Methods)] The manuscript asserts that an ensemble of independent runs at stepped, fixed bias strengths supplies the identical CV-efficiency information previously obtained from a continuously time-varying bias, allowing reuse of the original gamma formula. No derivation is supplied showing why the response of the observed rate (or non-equilibrium flux) to bias magnitude remains linear enough for the same correction to apply; the numerical agreement reported for the two test systems does not rule out protocol-dependent non-linear effects in transition-path sampling.
minor comments (2)
- [Abstract] The abstract states that the method is 'accurate without substantial loss of efficiency' but does not quantify what 'substantial' means or report the precise efficiency ratios and error bars for the two systems.
- [Results / Implementation] The internal over-biasing check is listed as an appealing feature but its implementation and decision criterion are not described in sufficient detail for a reader to reproduce or apply it.
Simulated Author's Rebuttal
We thank the referee for their constructive review and positive assessment of the significance and reproducibility of EATR-flooding. We address the major comment below and have revised the manuscript to provide additional justification for the generalization.
read point-by-point responses
-
Referee: The manuscript asserts that an ensemble of independent runs at stepped, fixed bias strengths supplies the identical CV-efficiency information previously obtained from a continuously time-varying bias, allowing reuse of the original gamma formula. No derivation is supplied showing why the response of the observed rate (or non-equilibrium flux) to bias magnitude remains linear enough for the same correction to apply; the numerical agreement reported for the two test systems does not rule out protocol-dependent non-linear effects in transition-path sampling.
Authors: We thank the referee for this important observation. The original EATR extracts gamma from the dependence of the observed rate on the instantaneous bias strength as the bias varies continuously with time. In EATR-flooding we instead obtain the same information by performing separate equilibrium simulations at a discrete ladder of fixed bias strengths; the resulting set of (bias strength, observed rate) pairs is then fitted to the same functional form. Because the underlying non-equilibrium flux expression depends on the instantaneous bias magnitude rather than on its time derivative, the linearity assumption with respect to bias strength carries over directly, and gamma remains a property of the CV alone. We have added a short derivation in the revised Methods section that starts from the biased flux and shows why the same gamma correction applies when bias strength is varied across independent runs rather than over time. We agree that numerical agreement on two systems does not constitute a general proof against all possible non-linearities; however, the internal over-biasing diagnostic now included in the library is intended to flag such deviations in practice, and the fact that a single gamma per CV is recovered (independent of the particular stepping schedule) already removes one source of protocol dependence that existed in the original EATR. We have updated the text to make these points explicit. revision: yes
Circularity Check
EATR-flooding generalization adds independent procedural elements without reducing to self-defined inputs
full rationale
The paper's central derivation replaces continuous time-dependent biasing in the prior EATR method with stepped bias strengths across independent simulation sets, while retaining the gamma correction and adding an internal over-biasing check. This is presented as a direct generalization with numerical validation on a coarse-grained protein and an atomistic cavity-ligand system. No equations or claims in the provided text reduce the new rate estimates or gamma by construction to quantities defined solely from the original EATR inputs or fits; the self-reference to prior EATR work supplies the base method but is not load-bearing for the flooding variant's validity, which rests on the new protocol and empirical tests. The derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- gamma
axioms (1)
- domain assumption Varying bias strength across independent runs captures the same efficiency information as continuous time variation of the bias.
Reference graph
Works this paper leans on
-
[1]
Binding kinetics in drug discovery
Ferruz, N.; De Fabritiis, G. Binding kinetics in drug discovery. Mol. Inform. 2016, 35, 216--226
2016
-
[2]
D.; Dickson, A
Lotz, S. D.; Dickson, A. Unbiased molecular dynamics of 11 min timescale drug unbinding reveals transition state stabilizing interactions. J. Am. Chem. Soc. 2018, 140, 618--628
2018
-
[3]
Kinetics of drug binding and residence time
Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of drug binding and residence time. Annu. Rev. Phys. Chem. 2019, 70, 143--171
2019
-
[4]
B.; Wade, R
Nunes-Alves, A.; Kokh, D. B.; Wade, R. C. Recent progress in molecular simulation methods for drug binding kinetics. Curr. Opin. Struct. Biol. 2020, 64, 126--133
2020
-
[5]
A.; Amaro, R
Ahn, S.-H.; Ojha, A. A.; Amaro, R. E.; McCammon, J. A. Gaussian-accelerated molecular dynamics with the weighted ensemble method: A hybrid method improves thermodynamic and kinetic sampling. J. Chem. Theory Comput. 2021, 17, 7938--7951
2021
-
[6]
Toward automated physics-based absolute drug residence time predictions
Smith, Z.; Branduardi, D.; Lupyan, D.; D’Arrigo, G.; Tiwary, P.; Wang, L.; Krilov, G. Toward automated physics-based absolute drug residence time predictions. J. Chem. Inf. Model. 2025, 65, 13360--13373
2025
-
[7]
Karplus, M.; McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646--652
2002
-
[8]
G.; Beljak, L.; Chen, J.; Dakhel, S.; Darling, D.; Ghosh, S.; Hall, J.; Jan, M.; Liang, E.; Saju, S.; Vohr, M.; Wu, C.; Xu, Y.; Xue, E
Schlick, T.; Portillo-Ledesma, S.; Myers, C. G.; Beljak, L.; Chen, J.; Dakhel, S.; Darling, D.; Ghosh, S.; Hall, J.; Jan, M.; Liang, E.; Saju, S.; Vohr, M.; Wu, C.; Xu, Y.; Xue, E. Biomolecular Modeling and Simulation: A Prospering Multidisciplinary Field. Ann. Rev. Biophys. 2021, 50, 267--301, PMID: 33606945
2021
-
[9]
Tuckerman, M. E. Statistical mechanics: theory and molecular simulation; Oxford university press, 2023
2023
-
[10]
Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1
H \'e nin, J.; Leli \`e vre, T.; Shirts, M.; Valsson, O.; Delemotte, L. Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1. 0]. LiveCoMS 2022, 4, 1583--1583
2022
-
[11]
Umbrella sampling
K \"a stner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932--942
2011
-
[12]
Escaping free-energy minima
Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562--12566
2002
-
[13]
Well-tempered metadynamics: a smoothly converging and tunable free-energy method
Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603
2008
-
[14]
Rethinking metadynamics: From bias potentials to probability distributions
Invernizzi, M.; Parrinello, M. Rethinking metadynamics: From bias potentials to probability distributions. J. Phys. Chem. Lett. 2020, 11, 2731--2736
2020
-
[15]
Variational approach to enhanced sampling and free energy calculations
Valsson, O.; Parrinello, M. Variational approach to enhanced sampling and free energy calculations. Phys. Rev. Lett. 2014, 113, 090601
2014
-
[16]
Using metadynamics to explore complex free-energy landscapes
Bussi, G.; Laio, A. Using metadynamics to explore complex free-energy landscapes. Nat. Rev. Phys. 2020, 2, 200--212
2020
-
[17]
Voter, A. F. Hyperdynamics: Accelerated molecular dynamics of infrequent events. Phys. Rev. Lett. 1997, 78, 3908
1997
-
[18]
From Metadynamics to Dynamics
Tiwary, P.; Parrinello, M. From Metadynamics to Dynamics. Phys. Rev. Lett. 2013, 111, 230602
2013
-
[19]
Assessing the Reliability of the Dynamics Reconstructed from Metadynamics
Salvalaglio, M.; Tiwary, P.; Parrinello, M. Assessing the Reliability of the Dynamics Reconstructed from Metadynamics. J. Chem. Theory Comput. 2014, 10, 1420--1425
2014
-
[20]
Rare event kinetics from adaptive bias enhanced sampling
Ray, D.; Ansari, N.; Rizzi, V.; Invernizzi, M.; Parrinello, M. Rare event kinetics from adaptive bias enhanced sampling. J. Chem. Theor. Comput. 2022, 18, 6500--6509
2022
-
[21]
Kinetics from Metadynamics: Principles, Applications, and Outlook
Ray, D.; Parrinello, M. Kinetics from Metadynamics: Principles, Applications, and Outlook. J. Chem. Theor. Comput. 2023, 19, 5649--5670
2023
-
[22]
Short-Time Infrequent Metadynamics for Improved Kinetics Inference
Blumer, O.; Reuveni, S.; Hirshberg, B. Short-Time Infrequent Metadynamics for Improved Kinetics Inference. J. Chem. Theor. Comput. 2024, 20, 3484--3491, PMID: 38668722
2024
-
[23]
S.; Pietrucci, F.; Hummer, G.; Cossio, P
Palacio-Rodriguez, K.; Vroylandt, H.; Stelzl, L. S.; Pietrucci, F.; Hummer, G.; Cossio, P. Transition Rates and Efficiency of Collective Variables from Time-Dependent Biased Simulations. J. Phys. Chem. Lett. 2022, 13, 7490--7496
2022
-
[24]
Mazzaferro, N.; Sasmal, S.; Cossio, P.; Hocky, G. M. Good Rates From Bad Coordinates: The Exponential Average Time-dependent Rate Approach. J. Chem. Theory Comput. 2024, 20, 5901--5912, PMID: 38954555
2024
-
[25]
Predicting slow structural transitions in macromolecular systems: Conformational flooding
Grubm \"u ller, H. Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys. Rev. E 1995, 52, 2893
1995
-
[26]
A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G
Tribello, G. A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Comm. 2014, 185, 604--613
2014
-
[27]
A.; Ban \'a s , P.; Barducci, A.; Bernetti, M.; Bolhuis, P
Bonomi, M.; Bussi, G.; Camilloni, C.; Tribello, G. A.; Ban \'a s , P.; Barducci, A.; Bernetti, M.; Bolhuis, P. G.; Bottaro, S.; Branduardi, D., et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670--673
2019
-
[28]
Voter, A. F. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys. 1997, 106, 4665--4677
1997
-
[29]
K.; Graham, T
Dudko, O. K.; Graham, T. G. W.; Best, R. B. Locating the Barrier for Folding of Single Molecules under an External Force. Phys. Rev. Lett. 2011, 107, 208301
2011
-
[30]
Graham, T. G. W.; Best, R. B. Force-Induced Change in Protein Unfolding Mechanism: Discrete or Continuous Switch? J. Phys. Chem. B 2011, 115, 1546--1561
2011
-
[31]
Mazzaferro, N.; Lee, S.; Cossio, P.; Tiwary, P.; Hocky, G. M. Using Time Dependent Rate Analysis to Evaluate the Quality of Machine Learned Reaction Coordinates for Biasing and Computing Kinetics. J. Phys. Chem. B 2025, 129, 10967--10974
2025
-
[32]
A.; Berne, B
Mondal, J.; Morrone, J. A.; Berne, B. J. How hydrophobic drying forces impact the kinetics of molecular recognition. Proc. Natl. Acad. Sci. USA 2013, 110, 13277--13282
2013
-
[33]
A.; Berne, B
Tiwary, P.; Mondal, J.; Morrone, J. A.; Berne, B. J. Role of water and steric constraints in the kinetics of cavity–ligand unbinding. Proc. Natl. Acad. Sci. USA 2015, 112, 12015--12019
2015
-
[34]
J.; Hocky, G
Pe\ n a Ccoa, W. J.; Hocky, G. M. Assessing models of force-dependent unbinding rates via infrequent metadynamics. J. Chem. Phys. 2022, 156, 125102
2022
-
[35]
On the role of solvent in hydrophobic cavity–ligand recognition kinetics
Ahalawat, N.; Bandyopadhyay, S.; Mondal, J. On the role of solvent in hydrophobic cavity–ligand recognition kinetics. J. Chem. Phys. 2020, 152, 074104
2020
-
[36]
J.; Mukadum, F.; Ramon, A.; Stirnemann, G.; Hocky, G
Pe \ n a Ccoa, W. J.; Mukadum, F.; Ramon, A.; Stirnemann, G.; Hocky, G. M. A direct computational assessment of vinculin--actin unbinding kinetics reveals catch-bonding behavior. Proc. Natl. Acad. Sci. USA 2025, 122, e2425982122
2025
-
[37]
P.; Aktulga, H
Thompson, A. P.; Aktulga, H. M.; Berger, R.; Bolintineanu, D. S.; Brown, W. M.; Crozier, P. S.; In't Veld, P. J.; Kohlmeyer, A.; Moore, S. G.; Nguyen, T. D., et al. LAMMPS-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Comm. 2022, 271, 108171
2022
-
[38]
J.; Murtola, T.; Schulz, R.; P \'a ll, S.; Smith, J
Abraham, M. J.; Murtola, T.; Schulz, R.; P \'a ll, S.; Smith, J. C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19--25
2015
-
[39]
A.; Bonomi, M.; Bussi, G.; Camilloni, C.; Armstrong, B
Tribello, G. A.; Bonomi, M.; Bussi, G.; Camilloni, C.; Armstrong, B. I.; Arsiccio, A.; Aureli, S.; Ballabio, F.; Bernetti, M.; Bonati, L., et al. PLUMED Tutorials: A collaborative, community-driven learning ecosystem. J. Chem. Phys. 2025, 162
2025
-
[40]
E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; van der Walt , S
Virtanen, P.; Gommers, R.; Oliphant, T. E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; van der Walt , S. J.; Brett, M.; Wilson, J.; Millman, K. J.; Mayorov, N.; Nelson, A. R. J.; Jones, E.; Kern, R.; Larson, E.; Carey, C. J.; Polat, \.I .; Feng, Y.; Moore, E. W.; VanderPlas , J.; Laxalde, D.; Perktold,...
2020
-
[41]
Bootstrap Methods: Another Look at the Jackknife
Efron, B. Bootstrap Methods: Another Look at the Jackknife. Ann. Stat. 1979, 7, 1 -- 26
1979
-
[42]
P j*j U 3˶ ysWsv zxm?߮ m8L_|e ŶR<mY=IJz7y< 0' ۳ x1枖ONk Kϯ|^ f <q? @ zGn |mg A
Ibrahim, J. G.; Chen, M.-H.; Sinha, D. Bayesian survival analysis; Springer Science & Business Media, 2001 mcitethebibliography eatr-flooding-plusSI-arxiv.tex0000664000000000000000000020453215174456253015420 0ustar rootroot [journal=jctcce,layout=twocolumn,manuscript=article] achemso acs articletitle = true amsmath bm dcolumn d [1] D . . #1 graphicx multi...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.