Recognition: unknown
Relativistic Exact-Two-Component Core-Valence-Separated Algebraic Diagrammatic Construction Theory For Near L-edge X-ray Absorption Spectra
Pith reviewed 2026-05-07 08:28 UTC · model grok-4.3
The pith
A two-component relativistic CVS-ADC(2) method with state-averaged frozen natural spinors and Cholesky decomposition reproduces four-component accuracy for L-edge X-ray spectra at reduced cost.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that the core-valence-separated algebraic diagrammatic construction at second order, implemented in the exact two-component (X2CMP/X2CAMF) relativistic framework and accelerated by state-averaged frozen natural spinors together with Cholesky decomposition, delivers results for near L-edge X-ray absorption spectra that closely match four-component reference calculations while requiring only a fraction of the computational cost. Benchmark studies on 3d transition-metal compounds confirm that this CVS-ADC(2) variant reproduces experimental L2,3-edge spectra with accuracy comparable to non-Hermitian EOM-CC, and the approach scales to medium-sized relativistic molecular cases
What carries the argument
State-averaged frozen natural spinors (SA-FNS) combined with Cholesky decomposition (CD) applied inside the two-component relativistic core-valence-separated ADC(2) equations to reduce floating-point operations and integral storage for core excitations.
If this is right
- The method serves as a reliable and computationally efficient alternative to four-component calculations for near L-edge spectra in systems containing heavy elements.
- CVS-ADC(2) reproduces experimental L2,3-edge spectra for 3d transition-metal compounds with accuracy comparable to non-Hermitian EOM-CC at lower cost.
- The framework extends to relativistic studies of medium-sized molecular systems such as ruthenium complexes.
- Systematic benchmarking confirms that the two-component (X2CMP/X2CAMF) framework maintains close agreement with canonical four-component results.
Where Pith is reading between the lines
- The efficiency gains could enable X-ray absorption calculations on larger molecules or materials with heavy atoms that exceed the reach of four-component methods.
- The approach could be paired with local correlation techniques or basis-set extrapolation to reach even bigger systems while retaining core-excitation accuracy.
- Additional comparisons on diverse heavy-element compounds would quantify any residual errors from the two-component approximation beyond the current benchmarks.
Load-bearing premise
The two-component relativistic approximation combined with SA-FNS truncation and Cholesky decomposition preserves sufficient accuracy for L-edge core excitations in the tested heavy-element systems without introducing systematic errors.
What would settle it
Substantial deviations between the two-component SA-FNS CVS-ADC(2) L-edge results and both four-component reference values and experimental spectra on an additional heavy-element molecule outside the current test set would falsify the reliability claim.
Figures



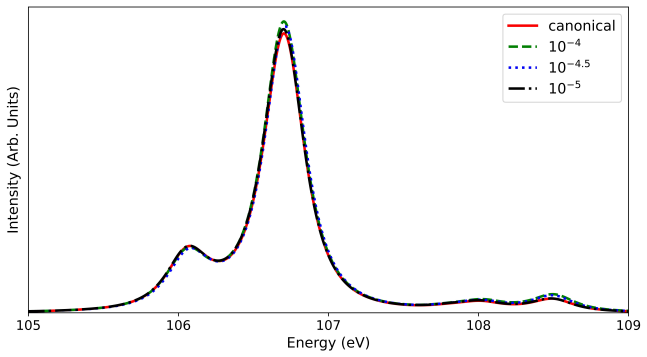
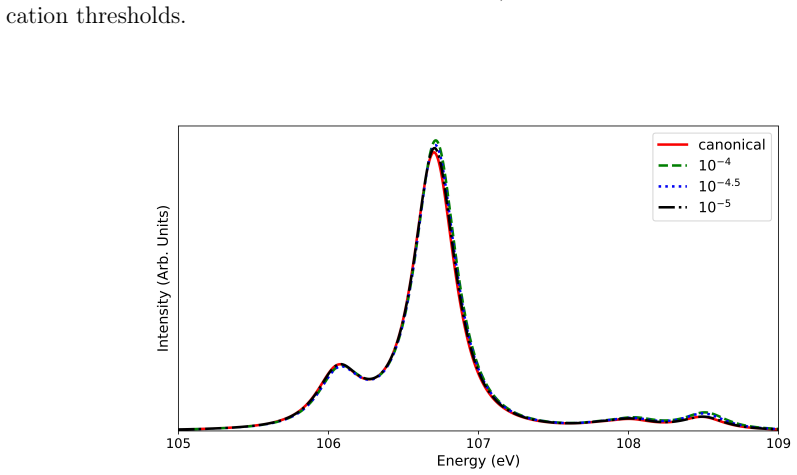




read the original abstract
We present an efficient implementation of the second-order two-component relativistic core-valence-separated algebraic diagrammatic construction method (CVS-ADC(2)) for core-excitation calculations. The approach employs state-averaged frozen natural spinors (SA-FNS) to reduce the number of floating-point operations, together with the Cholesky decomposition (CD) technique, which lowers the storage requirements associated with two-electron integrals. These reductions make the method particularly well-suited for systems containing heavy elements. Systematic benchmarking against four-component reference calculations confirms the reliability and robustness of the two-component (X2CMP/X2CAMF)-based framework. The close agreement with canonical results further demonstrates that the SA-FNS-based CVS-ADC(2) approach achieves comparable accuracy at only a fraction of the computational cost. Moreover, benchmark studies of L$_{2,3}$-edge spectra for 3$d$ transition-metal compounds demonstrate that CVS-ADC(2) serves as a computationally efficient and reliable alternative to the non-Hermitian EOM-CC method for reproducing experimental spectra. Finally, calculations on a ruthenium complex illustrate the method's applicability to relativistic studies of medium-sized molecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents an efficient implementation of the second-order core-valence-separated algebraic diagrammatic construction method (CVS-ADC(2)) within a two-component relativistic exact-two-component (X2C) framework (X2CMP/X2CAMF) for near L-edge X-ray absorption spectra. It employs state-averaged frozen natural spinors (SA-FNS) to truncate the virtual space and Cholesky decomposition (CD) to reduce integral storage, enabling calculations on heavy-element systems. Systematic benchmarks against four-component reference calculations and experimental L_{2,3}-edge spectra for 3d transition-metal compounds and a ruthenium complex are reported, with claims of close agreement, comparable accuracy to non-Hermitian EOM-CC, and substantial computational savings.
Significance. If the accuracy claims hold, the work provides a practical, scalable route to relativistic core-excitation spectra for medium-sized molecules containing heavy atoms, where four-component methods remain prohibitive. The SA-FNS + CD combination directly tackles the dominant computational costs in ADC(2) while retaining the core-valence separation, potentially enabling routine studies of L-edge XAS in 3d and 4d metal complexes that are currently limited to smaller systems or lower-level methods.
major comments (3)
- [Abstract / Benchmarking results] Abstract and benchmarking sections: The claims of 'systematic benchmarking against four-component reference calculations' and 'close agreement with canonical results' are not supported by quantitative error metrics (e.g., mean absolute deviations or maximum errors in excitation energies/intensities), the number or sizes of tested systems, or explicit values of the SA-FNS occupation threshold and CD tolerance. Without these, the assertion that the two-component framework 'preserves sufficient accuracy' for L-edge spectra cannot be evaluated.
- [Methodology (SA-FNS)] Methodology section describing SA-FNS: State averaging over frozen natural spinors is used to select a reduced virtual space, but for 2p→3d L_{2,3}-edge transitions the dominant 3d virtuals may receive diluted natural occupations when lower-lying valence states are included in the averaging. The manuscript must specify the occupation threshold, the states included in the average, and provide sensitivity tests (e.g., comparison to untruncated virtual space or varying thresholds) to demonstrate that no systematic shifts are introduced relative to the four-component references.
- [Results (L-edge benchmarks)] Results section on L_{2,3}-edge spectra for 3d compounds: The statement that CVS-ADC(2) 'serves as a computationally efficient and reliable alternative to the non-Hermitian EOM-CC method' requires explicit per-system comparisons, including which molecules were tested, the magnitude of deviations from experiment or EOM-CC, and any post-hoc adjustments. The current description supplies no such statistics, weakening the cross-method reliability claim.
minor comments (2)
- [Introduction] Define all acronyms (CVS-ADC(2), SA-FNS, X2CMP, X2CAMF, CD) at first use in the introduction and ensure consistent usage thereafter.
- [Results / Computational details] In any tables or figures comparing spectra or timings, include explicit error statistics or scaling data to quantify the claimed 'fraction of the computational cost.'
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments, which have identified opportunities to strengthen the quantitative support for our claims. We address each major point below and will revise the manuscript accordingly to improve clarity and rigor.
read point-by-point responses
-
Referee: [Abstract / Benchmarking results] Abstract and benchmarking sections: The claims of 'systematic benchmarking against four-component reference calculations' and 'close agreement with canonical results' are not supported by quantitative error metrics (e.g., mean absolute deviations or maximum errors in excitation energies/intensities), the number or sizes of tested systems, or explicit values of the SA-FNS occupation threshold and CD tolerance. Without these, the assertion that the two-component framework 'preserves sufficient accuracy' for L-edge spectra cannot be evaluated.
Authors: We agree that explicit quantitative metrics would better substantiate the benchmarking claims. In the revised manuscript we will add a summary table reporting mean absolute deviations and maximum errors in both excitation energies and intensities relative to the four-component references. The table will also list the specific molecules tested (including their sizes) along with the SA-FNS occupation threshold and Cholesky decomposition tolerance employed. These additions will allow direct evaluation of the accuracy of the two-component framework. revision: yes
-
Referee: [Methodology (SA-FNS)] Methodology section describing SA-FNS: State averaging over frozen natural spinors is used to select a reduced virtual space, but for 2p→3d L_{2,3}-edge transitions the dominant 3d virtuals may receive diluted natural occupations when lower-lying valence states are included in the averaging. The manuscript must specify the occupation threshold, the states included in the average, and provide sensitivity tests (e.g., comparison to untruncated virtual space or varying thresholds) to demonstrate that no systematic shifts are introduced relative to the four-component references.
Authors: We acknowledge the potential concern about dilution of natural occupations for the relevant virtual orbitals. The revised manuscript will explicitly state the occupation threshold used for virtual-space truncation, describe the states included in the state averaging (the L-edge core-excited states of interest), and include sensitivity tests comparing results at the chosen threshold against both the untruncated virtual space and alternative thresholds. These tests will demonstrate that any shifts remain well below the intrinsic accuracy of the CVS-ADC(2) method and show no systematic bias relative to four-component references. revision: yes
-
Referee: [Results (L-edge benchmarks)] Results section on L_{2,3}-edge spectra for 3d compounds: The statement that CVS-ADC(2) 'serves as a computationally efficient and reliable alternative to the non-Hermitian EOM-CC method' requires explicit per-system comparisons, including which molecules were tested, the magnitude of deviations from experiment or EOM-CC, and any post-hoc adjustments. The current description supplies no such statistics, weakening the cross-method reliability claim.
Authors: We agree that per-system statistics are required to support the cross-method comparison. The revised manuscript will include a table listing all tested 3d transition-metal compounds and the ruthenium complex, together with CVS-ADC(2) excitation energies and intensities, the corresponding EOM-CC values, experimental references where available, and the resulting deviations. We will also confirm that no post-hoc adjustments were applied to the computed spectra. This will provide the quantitative basis for the reliability claim. revision: yes
Circularity Check
No circularity; claims rest on external four-component benchmarks and experiments
full rationale
The paper presents an implementation of CVS-ADC(2) in a two-component relativistic framework using SA-FNS truncation and Cholesky decomposition for efficiency. Its reliability claims are supported by direct comparisons to independent four-component reference calculations and experimental L-edge spectra for 3d transition-metal compounds, which serve as external validation rather than self-referential fits or derivations. No load-bearing steps in the provided text reduce by construction to the method's own inputs, fitted parameters renamed as predictions, or self-citation chains. The derivation chain follows standard ADC equations adapted to the relativistic and core-valence-separated context without tautological reductions.
Axiom & Free-Parameter Ledger
free parameters (2)
- SA-FNS occupation threshold
- Cholesky decomposition tolerance
axioms (2)
- domain assumption The exact two-component (X2C) relativistic Hamiltonian accurately reproduces four-component core-excitation energies for L-edges in the systems studied.
- domain assumption State-averaged frozen natural spinors preserve the essential correlation effects for core-valence excitations.
Reference graph
Works this paper leans on
-
[1]
High-Resolution X-ray Emission and X-ray Absorption Spectroscopy
de Groot, F. High-Resolution X-ray Emission and X-ray Absorption Spectroscopy. Chemical Reviews 2001, 101, 1779--1808, PMID: 11709999
2001
-
[2]
NEXAFS Spectroscopy; Springer Series in Surface Sciences; Springer Berlin Heidelberg: Berlin, Heidelberg, 1992; Vol
St \"o hr, J. NEXAFS Spectroscopy; Springer Series in Surface Sciences; Springer Berlin Heidelberg: Berlin, Heidelberg, 1992; Vol. 25
1992
-
[3]
Recent experimental and theoretical developments in time-resolved X-ray spectroscopies
Milne, C.; Penfold, T.; Chergui, M. Recent experimental and theoretical developments in time-resolved X-ray spectroscopies. Coordination Chemistry Reviews 2014, 277-278, 44--68, Following Chemical Structures using Synchrotron Radiation
2014
-
[4]
T.; Hirsch, K.; Klar, P.; Langenberg, A.; Lofink, F.; Richter, R.; Rittmann, J.; Vogel, M.; Zamudio-Bayer, V.; M\"oller, T.; Issendorff, B
Lau, J. T.; Hirsch, K.; Klar, P.; Langenberg, A.; Lofink, F.; Richter, R.; Rittmann, J.; Vogel, M.; Zamudio-Bayer, V.; M\"oller, T.; Issendorff, B. v. X-ray spectroscopy reveals high symmetry and electronic shell structure of transition-metal-doped silicon clusters. Phys. Rev. A 2009, 79, 053201
2009
-
[5]
A.; Martini, A.; Bugaev, A
Soldatov, M. A.; Martini, A.; Bugaev, A. L.; Pankin, I.; Medvedev, P. V.; Guda, A. A.; Aboraia, A. M.; Podkovyrina, Y. S.; Budnyk, A. P.; Soldatov, A. A.; Lamberti, C. The insights from X-ray absorption spectroscopy into the local atomic structure and chemical bonding of Metal–organic frameworks. Polyhedron 2018, 155, 232--253
2018
-
[6]
I.; Suljoti, E.; Garcia-Diez, R.; Lange, K
Engel, N.; Bokarev, S. I.; Suljoti, E.; Garcia-Diez, R.; Lange, K. M.; Atak, K.; Golnak, R.; Kothe, A.; Dantz, M.; K \"u hn, O.; Aziz, E. F. Chemical Bonding in Aqueous Ferrocyanide: Experimental and Theoretical X-ray Spectroscopic Study. The Journal of Physical Chemistry B 2014, 118, 1555--1563, PMID: 24450820
2014
-
[7]
State of the Art
Baker, M. L.; Mara, M. W.; Yan, J. J.; Hodgson, K. O.; Hedman, B.; Solomon, E. I. K- and L-edge X-ray absorption spectroscopy (XAS) and resonant inelastic X-ray scattering (RIXS) determination of differential orbital covalency (DOC) of transition metal sites. Coordination Chemistry Reviews 2017, 345, 182--208, Chemical Bonding: "State of the Art"
2017
-
[8]
Bounding [AnO2]2+ (An = U , Np) covalency by simulated O K-edge and An M-edge X-ray absorption near-edge spectroscopy
Stanistreet-Welsh, K.; Kerridge, A. Bounding [AnO2]2+ (An = U , Np) covalency by simulated O K-edge and An M-edge X-ray absorption near-edge spectroscopy. Phys. Chem. Chem. Phys. 2023, 25, 23753--23760
2023
-
[9]
Cressey, G.; Henderson, C. M. B.; van der Laan, G. Use of L-edge X-ray absorption spectroscopy to characterize multiple valence states of 3d transition metals; a new probe for mineralogical and geochemical research. Physics and Chemistry of Minerals 1993, 20, 111--119
1993
-
[10]
Study of oxidation states of the transition metals in a series of Prussian blue analogs using x-ray absorption near edge structure (XANES) spectroscopy
Adak, S.; Hartl, M.; Daemen, L.; Fohtung, E.; Nakotte, H. Study of oxidation states of the transition metals in a series of Prussian blue analogs using x-ray absorption near edge structure (XANES) spectroscopy. Journal of Electron Spectroscopy and Related Phenomena 2017, 214, 8--19
2017
-
[11]
Kubin, M. et al. Probing the oxidation state of transition metal complexes: a case study on how charge and spin densities determine Mn L-edge X-ray absorption energies. Chem. Sci. 2018, 9, 6813--6829
2018
-
[12]
K.; Maganas, D.; DeBeer, S.; Neese, F
Chantzis, A.; Kowalska, J. K.; Maganas, D.; DeBeer, S.; Neese, F. Ab Initio Wave Function-Based Determination of Element Specific Shifts for the Efficient Calculation of X-ray Absorption Spectra of Main Group Elements and First Row Transition Metals. Journal of Chemical Theory and Computation 2018, 14, 3686--3702, PMID: 29894196
2018
-
[13]
Simulating X-ray Spectroscopies and Calculating Core-Excited States of Molecules
Norman, P.; Dreuw, A. Simulating X-ray Spectroscopies and Calculating Core-Excited States of Molecules. Chemical Reviews 2018, 118, 7208--7248, PMID: 29894157
2018
-
[14]
Besley, N. A. Density Functional Theory Based Methods for the Calculation of X-ray Spectroscopy. Accounts of Chemical Research 2020, 53, 1306--1315, PMID: 32613827
2020
-
[15]
D.; Penfold, T
Rankine, C. D.; Penfold, T. J. Progress in the Theory of X-ray Spectroscopy: From Quantum Chemistry to Machine Learning and Ultrafast Dynamics. The Journal of Physical Chemistry A 2021, 125, 4276--4293, PMID: 33729774
2021
-
[16]
Besley, N. A. Modeling of the spectroscopy of core electrons with density functional theory. WIREs Computational Molecular Science 2021, 11, e1527
2021
-
[17]
E.; Vidal, M
Fransson, T.; Brumboiu, I. E.; Vidal, M. L.; Norman, P.; Coriani, S.; Dreuw, A. XABOOM: An X-ray Absorption Benchmark of Organic Molecules Based on Carbon, Nitrogen, and Oxygen 1s ^* Transitions. Journal of Chemical Theory and Computation 2021, 17, 1618--1637, PMID: 33544612
2021
-
[18]
Relativistic four-component static-exchange approximation for core-excitation processes in molecules
Ekstr\"om, U.; Norman, P.; Carravetta, V. Relativistic four-component static-exchange approximation for core-excitation processes in molecules. Phys. Rev. A 2006, 73, 022501
2006
-
[19]
Ikeno, H.; de Groot, F. M. F.; Tanaka, I. Ab-initio CI calculations for 3d transition metal L2,3 X-ray absorption spectra of TiCl4 and VOCl3. Journal of Physics: Conference Series 2009, 190, 012005
2009
-
[20]
X-ray absorption resonances near L2 , 3-edges from real-time propagation of the Dirac–Kohn–Sham density matrix
Kadek, M.; Konecny, L.; Gao, B.; Repisky, M.; Ruud, K. X-ray absorption resonances near L2 , 3-edges from real-time propagation of the Dirac–Kohn–Sham density matrix. Phys. Chem. Chem. Phys. 2015, 17, 22566--22570
2015
-
[21]
Fransson, T.; Burdakova, D.; Norman, P. K- and L-edge X-ray absorption spectrum calculations of closed-shell carbon , silicon , germanium , and sulfur compounds using damped four-component density functional response theory. Phys. Chem. Chem. Phys. 2016, 18, 13591--13603
2016
-
[22]
K.; Saue, T
South, C.; Shee, A.; Mukherjee, D.; Wilson, A. K.; Saue, T. 4-Component relativistic calculations of L3 ionization and excitations for the isoelectronic species UO22+ , OUN+ and UN2. Phys. Chem. Chem. Phys. 2016, 18, 21010--21023
2016
-
[23]
L.; Shee, A.; Coriani, S.; Severo Pereira Gomes, A
Halbert, L.; Vidal, M. L.; Shee, A.; Coriani, S.; Severo Pereira Gomes, A. Relativistic EOM-CCSD for Core-Excited and Core-Ionized State Energies Based on the Four-Component Dirac–Coulomb(−Gaunt) Hamiltonian. Journal of Chemical Theory and Computation 2021, 17, 3583--3598, PMID: 33944570
2021
-
[24]
Accurate X-ray Absorption Spectra near L- and M-Edges from Relativistic Four-Component Damped Response Time-Dependent Density Functional Theory
Konecny, L.; Vicha, J.; Komorovsky, S.; Ruud, K.; Repisky, M. Accurate X-ray Absorption Spectra near L- and M-Edges from Relativistic Four-Component Damped Response Time-Dependent Density Functional Theory. Inorganic Chemistry 2022, 61, 830--846, PMID: 34958215
2022
-
[25]
Hess, B. A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742--3748
1986
-
[26]
J.; Snijders, J
van Lenthe, E.; van Leeuwen, R.; Baerends, E. J.; Snijders, J. G. Relativistic regular two-component Hamiltonians. International Journal of Quantum Chemistry 1996, 57, 281--293
1996
-
[27]
Dyall, K. G. Interfacing relativistic and nonrelativistic methods. I. Normalized elimination of the small component in the modified Dirac equation. The Journal of Chemical Physics 1997, 106, 9618--9626
1997
-
[28]
A new relativistic theory: a relativistic scheme by eliminating small components (RESC)
Nakajima, T.; Hirao, K. A new relativistic theory: a relativistic scheme by eliminating small components (RESC). Chemical Physics Letters 1999, 302, 383--391
1999
-
[29]
Barysz, M.; Sadlej, A. J. Two-component methods of relativistic quantum chemistry: from the Douglas–Kroll approximation to the exact two-component formalism. Journal of Molecular Structure: THEOCHEM 2001, 573, 181--200
2001
-
[30]
Exact two-component Hamiltonians revisited
Liu, W.; Peng, D. Exact two-component Hamiltonians revisited. The Journal of Chemical Physics 2009, 131, 031104
2009
-
[31]
Relativistic Hamiltonians for Chemistry: A Primer
Saue, T. Relativistic Hamiltonians for Chemistry: A Primer. ChemPhysChem 2011, 12, 3077--3094
2011
-
[32]
Quasirelativistic theory equivalent to fully relativistic theory
Kutzelnigg, W.; Liu, W. Quasirelativistic theory equivalent to fully relativistic theory. The Journal of Chemical Physics 2005, 123, 241102
2005
-
[33]
An infinite-order two-component relativistic Hamiltonian by a simple one-step transformation
Iliaš, M.; Saue, T. An infinite-order two-component relativistic Hamiltonian by a simple one-step transformation. The Journal of Chemical Physics 2007, 126, 064102
2007
-
[34]
G.; Faegri, K
Dyall, K. G.; Faegri, K. Introduction to Relativistic Quantum Chemistry; Oxford University Press, 2007
2007
-
[35]
An atomic mean-field spin-orbit approach within exact two-component theory for a non-perturbative treatment of spin-orbit coupling
Liu, J.; Cheng, L. An atomic mean-field spin-orbit approach within exact two-component theory for a non-perturbative treatment of spin-orbit coupling. The Journal of Chemical Physics 2018, 148, 144108
2018
-
[36]
Atomic Mean-Field Approach within Exact Two-Component Theory Based on the Dirac-Coulomb-Breit Hamiltonian
Zhang, C.; Cheng, L. Atomic Mean-Field Approach within Exact Two-Component Theory Based on the Dirac-Coulomb-Breit Hamiltonian. The Journal of Physical Chemistry A 2022, 126, 4537--4553, PMID: 35763592
2022
-
[37]
Knecht, S.; Repisky, M.; Jensen, H. J. A.; Saue, T. Exact two-component Hamiltonians for relativistic quantum chemistry: Two-electron picture-change corrections made simple. The Journal of Chemical Physics 2022, 157, 114106
2022
-
[38]
Infinite-order quasirelativistic density functional method based on the exact matrix quasirelativistic theory
Liu, W.; Peng, D. Infinite-order quasirelativistic density functional method based on the exact matrix quasirelativistic theory. The Journal of Chemical Physics 2006, 125, 044102
2006
-
[39]
from atoms to molecule
Peng, D.; Liu, W.; Xiao, Y.; Cheng, L. Making four- and two-component relativistic density functional methods fully equivalent based on the idea of “from atoms to molecule”. The Journal of Chemical Physics 2007, 127, 104106
2007
-
[40]
Relativistic two-electron contributions within exact two-component theory
Wang, X.; Zhang, C.; Liu, J.; Cheng, L. Relativistic two-electron contributions within exact two-component theory. Chemical Physics Reviews 2025, 6, 031404
2025
-
[41]
M.; Lestrange, P
Kasper, J. M.; Lestrange, P. J.; Stetina, T. F.; Li, X. Modeling L2,3-Edge X-ray Absorption Spectroscopy with Real-Time Exact Two-Component Relativistic Time-Dependent Density Functional Theory. Journal of Chemical Theory and Computation 2018, 14, 1998--2006, PMID: 29561613
2018
-
[42]
F.; Kasper, J
Stetina, T. F.; Kasper, J. M.; Li, X. Modeling L2,3-edge X-ray absorption spectroscopy with linear response exact two-component relativistic time-dependent density functional theory. The Journal of Chemical Physics 2019, 150, 234103
2019
-
[43]
H.; Dunford, R
Southworth, S. H.; Dunford, R. W.; Ray, D.; Kanter, E. P.; Doumy, G.; March, A. M.; Ho, P. J.; Kr\"assig, B.; Gao, Y.; Lehmann, C. S.; Pic\'on, A.; Young, L.; Walko, D. A.; Cheng, L. Observing pre-edge K -shell resonances in Kr, Xe, and XeF _ 2 . Phys. Rev. A 2019, 100, 022507
2019
-
[44]
H.; Cheng, L
Zheng, X.; Zhang, C.; Jin, Z.; Southworth, S. H.; Cheng, L. Benchmark relativistic delta-coupled-cluster calculations of K-edge core-ionization energies of third-row elements. Phys. Chem. Chem. Phys. 2022, 24, 13587--13596
2022
-
[45]
H.; Doumy, G.; Young, L.; Cheng, L
Wu, Y.; Lin, Z.; Wang, X.; Wang, S.; Southworth, S. H.; Doumy, G.; Young, L.; Cheng, L. Relativistic core–valence-separated equation-of-motion coupled-cluster singles and doubles method: Efficient implementation and benchmark calculations. The Journal of Chemical Physics 2025, 163, 244114
2025
-
[46]
J.; Wilson, R
Wang, X.; Doumy, G.; Marie March, A.; Otolski, C. J.; Wilson, R. E.; Walko, D. A.; Cheng, L.; Southworth, S. H. Resonant x-ray emission across the L3 edge of uranium compounds. Journal of Physics B: Atomic, Molecular and Optical Physics 2025, 58, 045602
2025
-
[47]
R.; Cooper, B
Banerjee, S.; Li, R. R.; Cooper, B. C.; Zhang, T.; Valeev, E. F.; Li, X.; DePrince, I., A. Eugene Relativistic core–valence-separated molecular mean-field exact-two-component equation-of-motion coupled cluster theory: Applications to L-edge x-ray absorption spectroscopy. APL Computational Physics 2026, 2, 016101
2026
-
[48]
A.; Marian, C
Heß, B. A.; Marian, C. M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chemical Physics Letters 1996, 251, 365--371
1996
-
[49]
Exact Two-Component TDDFT with Simple Two-Electron Picture-Change Corrections: X-ray Absorption Spectra Near L- and M-Edges of Four-Component Quality at Two-Component Cost
Konecny, L.; Komorovsky, S.; Vicha, J.; Ruud, K.; Repisky, M. Exact Two-Component TDDFT with Simple Two-Electron Picture-Change Corrections: X-ray Absorption Spectra Near L- and M-Edges of Four-Component Quality at Two-Component Cost. The Journal of Physical Chemistry A 2023, 127, 1360--1376, PMID: 36722848
2023
-
[50]
Accurate Relativistic Real-Time Time-Dependent Density Functional Theory for Valence and Core Attosecond Transient Absorption Spectroscopy
Moitra, T.; Konecny, L.; Kadek, M.; Rubio, A.; Repisky, M. Accurate Relativistic Real-Time Time-Dependent Density Functional Theory for Valence and Core Attosecond Transient Absorption Spectroscopy. The Journal of Physical Chemistry Letters 2023, 14, 1714--1724, PMID: 36757216
2023
-
[51]
Analysis of self-interaction correction for describing core excited states
Imamura, Y.; Nakai, H. Analysis of self-interaction correction for describing core excited states. International Journal of Quantum Chemistry 2007, 107, 23--29
2007
-
[52]
J.; Nguyen, P
Lestrange, P. J.; Nguyen, P. D.; Li, X. Calibration of Energy-Specific TDDFT for Modeling K-edge XAS Spectra of Light Elements. Journal of Chemical Theory and Computation 2015, 11, 2994--2999, PMID: 26575736
2015
-
[53]
ROWE, D. J. Equations-of-Motion Method and the Extended Shell Model. Rev. Mod. Phys. 1968, 40, 153--166
1968
-
[54]
F.; Bartlett, R
Stanton, J. F.; Bartlett, R. J. The equation of motion coupled‐cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. The Journal of Chemical Physics 1993, 98, 7029--7039
1993
-
[55]
Nooijen, M.; Bartlett, R. J. Equation of motion coupled cluster method for electron attachment. The Journal of Chemical Physics 1995, 102, 3629--3647
1995
-
[56]
S.; Domcke, W.; Schirmer, J
Cederbaum, L. S.; Domcke, W.; Schirmer, J. Many-body theory of core holes. Phys. Rev. A 1980, 22, 206--222
1980
-
[57]
Communication: X-ray absorption spectra and core-ionization potentials within a core-valence separated coupled cluster framework
Coriani, S.; Koch, H. Communication: X-ray absorption spectra and core-ionization potentials within a core-valence separated coupled cluster framework. The Journal of Chemical Physics 2015, 143, 181103
2015
-
[58]
L.; Pokhilko, P.; Krylov, A
Vidal, M. L.; Pokhilko, P.; Krylov, A. I.; Coriani, S. Equation-of-Motion Coupled-Cluster Theory to Model L-Edge X-ray Absorption and Photoelectron Spectra. The Journal of Physical Chemistry Letters 2020, 11, 8314--8321, PMID: 32897075
2020
-
[59]
Relativistic coupled-cluster and equation-of-motion coupled-cluster methods
Liu, J.; Cheng, L. Relativistic coupled-cluster and equation-of-motion coupled-cluster methods. WIREs Computational Molecular Science 2021, 11, e1536
2021
-
[60]
Schirmer, J.; Trofimov, A. B. Intermediate state representation approach to physical properties of electronically excited molecules. The Journal of chemical physics 2004, 120, 11449--11464
2004
-
[61]
Algebraic propagator approaches and intermediate-state representations
Mertins, F.; Schirmer, J. Algebraic propagator approaches and intermediate-state representations. I. The biorthogonal and unitary coupled-cluster methods. Physical Review A 1996, 53, 2140
1996
-
[62]
Closed-form intermediate representations of many-body propagators and resolvent matrices
Schirmer, J. Closed-form intermediate representations of many-body propagators and resolvent matrices. Physical Review A 1991, 43, 4647
1991
-
[63]
Beyond the random-phase approximation: A new approximation scheme for the polarization propagator
Schirmer, J. Beyond the random-phase approximation: A new approximation scheme for the polarization propagator. Physical Review A 1982, 26, 2395
1982
-
[64]
von Niessen, W.; Schirmer, J.; Cederbaum, L. S. Computational methods for the one-particle green's function. Computer Physics Reports 1984, 1, 57--125
1984
-
[65]
The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states
Dreuw, A.; Wormit, M. The algebraic diagrammatic construction scheme for the polarization propagator for the calculation of excited states. Wiley Interdisciplinary Reviews: Computational Molecular Science 2015, 5, 82--95
2015
-
[66]
L.; Schneider, M.; Hodecker, M.; Dreuw, A
Dempwolff, A. L.; Schneider, M.; Hodecker, M.; Dreuw, A. Efficient implementation of the non-Dyson third-order algebraic diagrammatic construction approximation for the electron propagator for closed-and open-shell molecules. The Journal of Chemical Physics 2019, 150
2019
-
[67]
Banerjee, S.; Sokolov, A. Y. Efficient Implementation of the Single-Reference Algebraic Diagrammatic Construction Theory for Charged Excitations: Applications to the TEMPO Radical and DNA Base Pairs. The Journal of Chemical Physics 2021, 154, 074105
2021
-
[68]
Core-level electronic spectra in ADC (2) approximation for polarization propagator: Carbon monoxide and nitrogen molecules
Trofimov, A.; Moskovskaya, T.; Gromov, E.; Vitkovskaya, N.; Schirmer, J. Core-level electronic spectra in ADC (2) approximation for polarization propagator: Carbon monoxide and nitrogen molecules. Journal of Structural Chemistry 2000, 41, 483--494
2000
-
[69]
Calculating X-ray absorption spectra of open-shell molecules with the unrestricted algebraic-diagrammatic construction scheme for the polarization propagator
Wenzel, J.; Wormit, M.; Dreuw, A. Calculating X-ray absorption spectra of open-shell molecules with the unrestricted algebraic-diagrammatic construction scheme for the polarization propagator. Journal of chemical theory and computation 2014, 10, 4583--4598
2014
-
[70]
Wenzel, J.; Wormit, M.; Dreuw, A. Calculating core-level excitations and X-ray absorption spectra of medium-sized closed-shell molecules with the algebraic-diagrammatic construction scheme for the polarization propagator. Journal of computational chemistry 2014, 35, 1900--1915
2014
-
[71]
Simulating X-ray emission spectroscopy with algebraic diagrammatic construction schemes for the polarization propagator
Fransson, T.; Dreuw, A. Simulating X-ray emission spectroscopy with algebraic diagrammatic construction schemes for the polarization propagator. Journal of chemical theory and computation 2018, 15, 546--556
2018
-
[72]
Consistent third-order one-particle transition and excited-state properties within the algebraic-diagrammatic construction scheme for the polarization propagator
Maier, R.; Bauer, M.; Dreuw, A. Consistent third-order one-particle transition and excited-state properties within the algebraic-diagrammatic construction scheme for the polarization propagator. The Journal of Chemical Physics 2023, 159
2023
-
[73]
Scheurer, M.; Fransson, T.; Norman, P.; Dreuw, A.; Rehn, D. R. Complex excited state polarizabilities in the ADC/ISR framework. The Journal of Chemical Physics 2020, 153
2020
-
[74]
The relativistic polarization propagator for the calculation of electronic excitations in heavy systems
Pernpointner, M. The relativistic polarization propagator for the calculation of electronic excitations in heavy systems. The Journal of Chemical Physics 2014, 140
2014
-
[75]
Pernpointner, M.; Visscher, L.; Trofimov, A. B. Four-component polarization propagator calculations of electron excitations: Spectroscopic implications of spin--orbit coupling effects. Journal of Chemical Theory and Computation 2018, 14, 1510--1522
2018
-
[76]
The one-particle Green’s function method in the Dirac--Hartree--Fock framework
Pernpointner, M.; Trofimov, A. The one-particle Green’s function method in the Dirac--Hartree--Fock framework. I. Second-order valence ionization energies of Ne through Xe. The Journal of chemical physics 2004, 120, 4098--4106
2004
-
[77]
The four-component two-particle propagator for the calculation of double-ionization spectra of heavy-element compounds: I
Pernpointner, M. The four-component two-particle propagator for the calculation of double-ionization spectra of heavy-element compounds: I. Method. Journal of Physics B: Atomic, Molecular and Optical Physics 2010, 43, 205102
2010
-
[78]
The effect of the Gaunt interaction on the molecular ionization spectra of CO, H2S and TlH
Pernpointner, M. The effect of the Gaunt interaction on the molecular ionization spectra of CO, H2S and TlH. Journal of Physics B: Atomic, Molecular and Optical Physics 2005, 38, 1955
2005
-
[79]
Mandal, S.; Dutta, A. K. A third-order relativistic algebraic diagrammatic construction method for double ionization potentials: Theory, implementation, and benchmark. The Journal of Chemical Physics 2026, 164
2026
-
[80]
K.; Dutta, A
Chakraborty, S.; Mukhopadhyay, T.; Nayak, M. K.; Dutta, A. K. A relativistic third-order algebraic diagrammatic construction theory for electron detachment, attachment, and excitation problems. The Journal of Chemical Physics 2025, 162, 104106
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.