Fragment-Constrained Charge Equilibration for Charge-Aware Machine Learning Potentials at Electrochemical Interfaces
Pith reviewed 2026-05-07 06:23 UTC · model grok-4.3
The pith
A differentiable fragment-constrained charge solver lets machine-learned potentials maintain separate electrode and electrolyte electrochemical potentials at interfaces.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
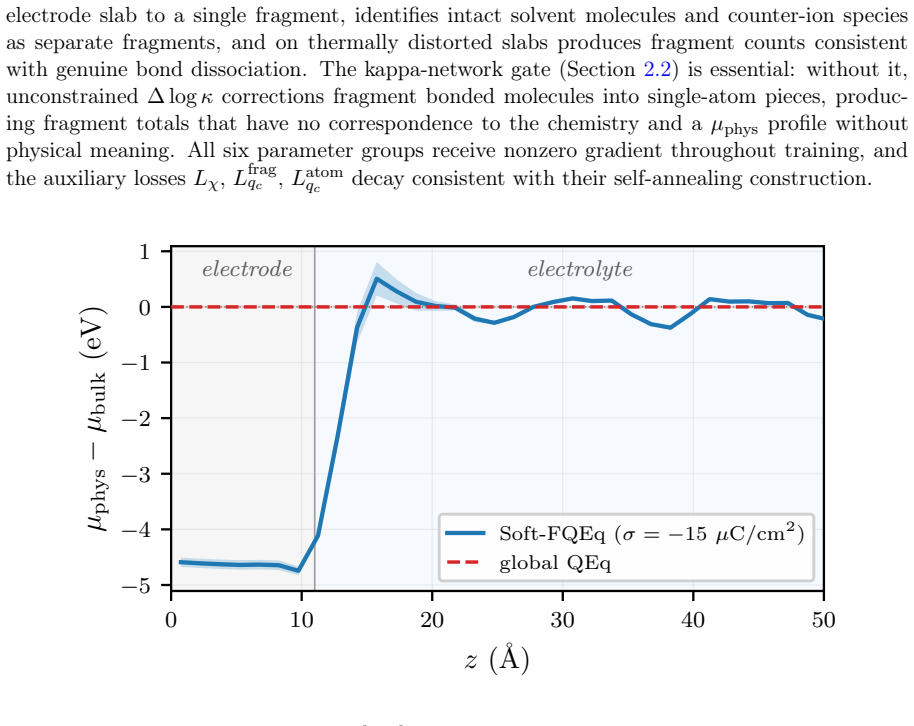
The paper establishes that a machine learning potential equipped with fragment-constrained charge equilibration recovers a clear electrode-to-electrolyte gradient in the per-atom electrochemical potential. The identical model weights, when evaluated with global charge equilibration instead, produce an essentially uniform potential profile. This contrast demonstrates that global equilibration cannot sustain the interfacial charge separation required for realistic double-layer behavior, whereas the fragment formulation restores it even in the presence of reactive chemistry.
What carries the argument
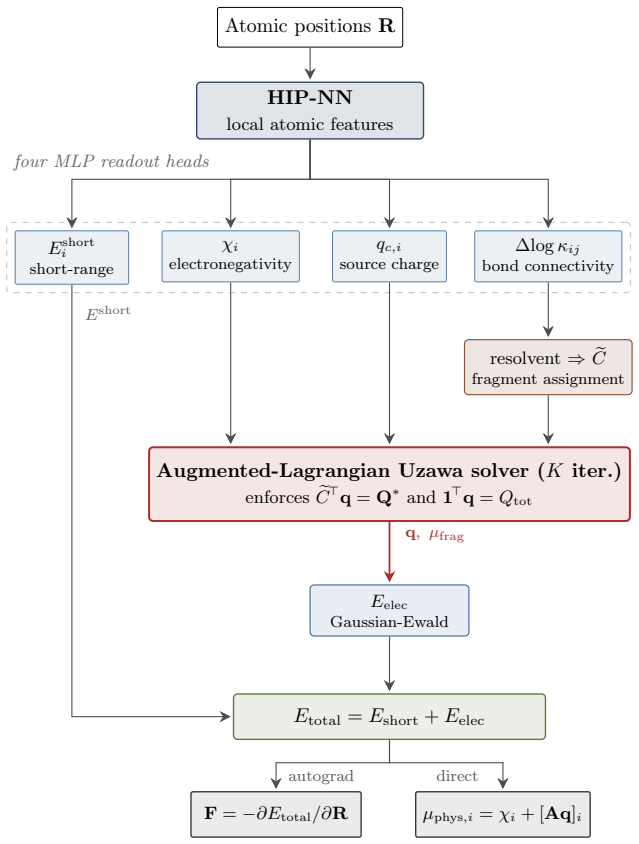
Soft-FQEq is a differentiable solver that ingests four scalar outputs per atom from a shared neural network feature extractor—electronegativity, source charge, short-range energy, and a soft bond-connectivity measure—and returns equilibrated charges plus distinct chemical potentials for each geometry-identified fragment.
If this is right
- Machine learning potentials can now simulate reactive bond rearrangements at electrochemical interfaces without allowing spurious charge transfer between electronically disconnected regions.
- Per-fragment chemical potentials become directly available from the model, providing a built-in diagnostic for local electrochemical driving forces.
- Training on combined density functional theory energies, forces, and atomic charges is now feasible for systems containing distinct electronic domains such as metal oxide electrodes in contact with aqueous electrolytes.
- Charge-aware potentials can be applied to dynamic, non-predefined topologies, extending their range to processes where molecular fragments change identity over time.
Where Pith is reading between the lines
- The geometry-only fragmentation rule may allow the same solver to be attached to other neural network architectures without requiring explicit molecular topology input.
- If the recovered gradient remains stable across varied training sets, the method could reduce reliance on separate continuum electrostatic models for interface simulations.
- Testing whether the soft connectivity continues to separate fragments correctly in organic electrolytes or during faradaic reactions would probe the assumption's generality beyond the presented oxide-water case.
- The per-fragment potentials could serve as local order parameters for enhanced sampling of charge-transfer events in larger-scale reactive simulations.
Load-bearing premise
A soft bond-connectivity function computed solely from atomic geometry can group atoms into fragments that correctly reflect the electronic separation between electrode and electrolyte regions, even while bonds rearrange during reactions.
What would settle it
Evaluating the trained model on held-out interface configurations and finding that the average per-atom electrochemical potential shows no statistically significant difference between electrode atoms and adjacent electrolyte atoms would falsify the claim that the fragment-constrained solver recovers the required gradient.
Figures


read the original abstract
Predictive simulation of electrochemical interfaces requires atomistic models that capture reactive bond rearrangements, long-range electrostatics, and charge distributions reflecting the electronic distinctness of electrode and electrolyte. Existing charge-aware machine-learned interatomic potentials (MLIPs) built on global charge equilibration (QEq) settle electrode and electrolyte at a common electrochemical potential, leaving no room for the interfacial gradient that the double layer requires and admitting spurious charge transfer between electronically disconnected regions. Per-fragment charge equilibration is the established remedy in classical molecular dynamics, but reliance on predefined molecular topology has confined it to non-reactive systems. We lift this restriction by making fragment identification itself a differentiable function of atomic geometry, yielding soft fragment-constrained charge equilibration (Soft-FQEq) -- a solver layer that restores fragment-resolved charge conservation in reactive MLIPs. The layer consumes four scalar MLP readouts from a shared atomic-feature network -- per-atom electronegativity, source charge, short-range energy, and a soft bond connectivity -- and returns equilibrated charges together with per-fragment chemical potentials. We implement Soft-FQEq as an extension of the hippynn framework on a HIP-NN feature network and train it on DFT energies, forces, and DDEC6 charges for IrO2/H2O/Na+/ClO4- interfaces. The trained model recovers a clear electrode-to-electrolyte gradient in the per-atom electrochemical potential. With the same trained weights but the fragment-constrained solver replaced by global QEq at inference, this gradient collapses to an essentially uniform profile, directly showing that the gradient cannot be sustained within global QEq while the fragment formulation recovers it.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces Soft-FQEq, a differentiable solver layer for fragment-constrained charge equilibration in charge-aware MLIPs. A shared HIP-NN feature network produces four scalar outputs per atom (electronegativity, source charge, short-range energy, and soft bond connectivity). These feed the Soft-FQEq solver to yield equilibrated charges and per-fragment chemical potentials while allowing reactive bond rearrangements. Trained on DFT energies, forces, and DDEC6 charges for IrO2/H2O/Na+/ClO4- interfaces, the model produces a clear electrode-to-electrolyte gradient in the per-atom electrochemical potential. Replacing the solver at inference with global QEq (identical MLP weights) causes the gradient to collapse to a uniform profile, which the authors interpret as direct evidence that global QEq cannot sustain the required interfacial potential difference.
Significance. If the learned soft connectivity reliably partitions the system into electronically distinct fragments, the work would be significant for electrochemical interface modeling. It removes the topology restriction that has limited per-fragment QEq to non-reactive classical MD, while retaining the ability to train end-to-end on DFT targets. The solver-swap experiment with fixed weights provides a clean isolation of the solver effect and directly addresses a known limitation of global-QEq MLIPs. The approach could enable ML-driven simulations that capture double-layer electrostatics without ad-hoc charge constraints.
major comments (3)
- [results section describing the per-atom electrochemical potential gradient] The central demonstration (abstract and results on the IrO2/H2O interface) shows recovery of the electrode-to-electrolyte gradient under Soft-FQEq and its collapse under global QEq. However, this contrast is only informative if the soft bond connectivity matrix defines fragments whose boundaries coincide with the physical electrode/electrolyte division. Because the connectivity is a learned, geometry-only function with no electronic-structure input and the training objective contains no explicit term enforcing electronic distinctness, nothing guarantees that the connected components respect the intended partition rather than forming mixed electrode–adsorbate clusters or splitting the electrode. Without visualizations of the connectivity matrix, statistics on cross-boundary bonds, or overlap metrics with known physical regions, the observed gradient could be an artifact of whatever ad-hoc
- [training and implementation details (hippynn extension)] The manuscript reports training on DFT energies, forces, and DDEC6 charges but provides no quantitative error metrics, training or validation curves, loss-component weights, or test-set performance for the IrO2/H2O/Na+/ClO4- system. In the absence of these data it is impossible to judge whether the MLP has converged to a solution that genuinely supports the solver comparison or whether the gradient recovery is sensitive to under-training or overfitting.
- [Soft-FQEq solver layer description] The soft bond connectivity is described as a differentiable function of atomic geometry with a single free softness parameter. No analysis is given of how this parameter is chosen or regularized, nor of its sensitivity: small changes could alter fragment boundaries and thereby change the recovered chemical-potential gradient. Because the gradient is the headline observable, this parameter choice is load-bearing and requires explicit validation.
minor comments (2)
- Clarify the precise mathematical definition of the soft bond connectivity readout (including the functional form, cutoff, and how it is normalized into a connectivity matrix) so that the solver layer can be reproduced independently.
- The abstract states that the model 'recovers a clear electrode-to-electrolyte gradient'; a quantitative measure (e.g., slope or potential difference across the interface) would strengthen the claim and allow direct comparison with global QEq.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments, which identify key areas where additional evidence and documentation will strengthen the manuscript. We address each major point below and commit to the indicated revisions.
read point-by-point responses
-
Referee: The central demonstration (abstract and results on the IrO2/H2O interface) shows recovery of the electrode-to-electrolyte gradient under Soft-FQEq and its collapse under global QEq. However, this contrast is only informative if the soft bond connectivity matrix defines fragments whose boundaries coincide with the physical electrode/electrolyte division. Because the connectivity is a learned, geometry-only function with no electronic-structure input and the training objective contains no explicit term enforcing electronic distinctness, nothing guarantees that the connected components respect the intended partition rather than forming mixed electrode–adsorbate clusters or splitting the electrode. Without visualizations of the connectivity matrix, statistics on cross-boundary bonds, or overlap metrics with known physical regions, the observed gradient could be an artifact of whatever ad-hoc
Authors: We agree that explicit validation of the learned fragment boundaries is essential to rule out artifacts. Although training on DDEC6 charges (which reflect physical charge distributions) provides implicit guidance toward appropriate partitions, we will add in the revision: (i) visualizations of the soft connectivity matrix for representative interface snapshots, (ii) statistics on the fraction of cross-boundary soft bonds, and (iii) overlap metrics between the connected components and the known physical electrode/electrolyte regions. These additions will confirm that the fragments align with the physical division and that the gradient recovery is not an artifact of the solver-swap experiment. revision: yes
-
Referee: The manuscript reports training on DFT energies, forces, and DDEC6 charges but provides no quantitative error metrics, training or validation curves, loss-component weights, or test-set performance for the IrO2/H2O/Na+/ClO4- system. In the absence of these data it is impossible to judge whether the MLP has converged to a solution that genuinely supports the solver comparison or whether the gradient recovery is sensitive to under-training or overfitting.
Authors: We acknowledge that these quantitative details were omitted from the original submission. In the revised manuscript we will add a dedicated subsection (or supplementary material) containing: mean absolute errors for energies, forces, and charges on training/validation/test splits; training and validation loss curves; the relative weights assigned to each loss term; and confirmation that the model converged without overfitting. This information will allow readers to assess the quality of the underlying MLP and the reliability of the solver comparison. revision: yes
-
Referee: The soft bond connectivity is described as a differentiable function of atomic geometry with a single free softness parameter. No analysis is given of how this parameter is chosen or regularized, nor of its sensitivity: small changes could alter fragment boundaries and thereby change the recovered chemical-potential gradient. Because the gradient is the headline observable, this parameter choice is load-bearing and requires explicit validation.
Authors: The softness parameter was selected to maintain differentiability while producing sufficiently distinct fragments; a small value was used to sharpen boundaries. To address the referee’s concern we will include in the revision a sensitivity analysis that varies the parameter over a physically reasonable range and reports the resulting changes (or stability) in the electrochemical potential gradient. We will also document the selection criterion and any regularization applied during training. revision: yes
Circularity Check
No circularity: central result follows from external DFT training targets and inference-time solver swap
full rationale
The paper trains the MLP end-to-end with the differentiable Soft-FQEq solver on independent DFT energies, forces, and DDEC6 charges. The headline demonstration applies the identical trained weights once with the fragment-constrained solver (recovering the electrode-electrolyte gradient in per-fragment chemical potentials) and once with global QEq (yielding a uniform profile). This contrast is not forced by construction: the training objective does not presuppose the gradient, the soft connectivity is optimized to reproduce external charge distributions rather than being defined to enforce electrode-electrolyte separation, and no self-citation, uniqueness theorem, or ansatz is invoked to justify the observed difference. The derivation chain remains self-contained against the external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (1)
- softness parameter for bond connectivity
axioms (2)
- domain assumption Charge must be conserved independently within each geometry-defined fragment.
- ad hoc to paper A differentiable function of atomic positions can identify fragments that correspond to electronically distinct regions.
invented entities (1)
-
Soft bond connectivity readout
no independent evidence
Reference graph
Works this paper leans on
-
[1]
J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes, and J. K. Nørskov. Electrolysis of water on oxide surfaces.Journal of Electroanalytical Chemistry, 607:83–89, 2007
work page 2007
-
[2]
Rik V. Mom, Lorenz J. Falling, Olga Kasian, Gerardo Algara-Siller, Detre Teschner, Robert H. Crabtree, Axel Knop-Gericke, Karl J. J. Mayrhofer, Juan-Jesús Velasco-Vélez, and Travis E. Jones. Operando structure–activity–stability relationship of iridium oxides during the oxygen evolution reaction.ACS Catalysis, 12(9):5174–5184, 2022
work page 2022
-
[3]
Xiongwei Tian, Axel Tosello Gardini, Umberto Raucci, Hai Xiao, Yuqun Zhuo, and Michele Parrinello. Electrochemical potential-driven water dynamics control CO2 electroreduction at the Ag/H2O interface.Nature Communications, 16:10636, 2025
work page 2025
-
[4]
Richard Schmuch, Ralf Wagner, Gerhard Hörpel, Tobias Placke, and Martin Winter. Per- formance and cost of materials for lithium-based rechargeable automotive batteries.Nature Energy, 3:267–278, 2018
work page 2018
-
[5]
Grey, Bruce Dunn, and Patrice Simon
Mathieu Salanne, Benjamin Rotenberg, Katsuhiko Naoi, Katsumi Kaneko, Pierre-Louis Taberna, Clare P. Grey, Bruce Dunn, and Patrice Simon. Efficient storage mechanisms for building better supercapacitors.Nature Energy, 1:16070, 2016
work page 2016
-
[6]
Chen Shen, Siamak Attarian, Yixuan Zhang, Hongbin Zhang, Mark Asta, Izabela Szlu- farska, and Dane Morgan. SuperSalt: Equivariant neural network force fields for multicom- ponent molten salts system.Nature Communications, 16:7280, 2025
work page 2025
-
[7]
Local reaction environment in electrocatalysis.Chemical Society Reviews, 53:2022–2055, 2024
Chaojie Chen, Huanyu Jin, Pengtang Wang, Xiaogang Sun, Mietek Jaroniec, Yao Zheng, and Shi-Zhang Qiao. Local reaction environment in electrocatalysis.Chemical Society Reviews, 53:2022–2055, 2024
work page 2022
-
[8]
Gebbie, Beichen Liu, Wenxiao Guo, Seth R
Matthew A. Gebbie, Beichen Liu, Wenxiao Guo, Seth R. Anderson, and Samuel G. John- stone. Linking electric double layer formation to electrocatalytic activity.ACS Catalysis, 13:16222–16239, 2023
work page 2023
-
[9]
Jianzhong Wu. Understanding the electric double-layer structure, capacitance, and charging dynamics.Chemical Reviews, 122(12):10821–10859, 2022
work page 2022
-
[10]
Zhuoran Long, Jinhui Meng, Lydia R. Weddle, Pablo E. Videla, Jan Paul Menzel, Delmar G. A. Cabral, Jinchan Liu, Tianyin Qiu, Joseph M. Palasz, Dhritiman Bhattacharyya, Clifford P. Kubiak, Victor S. Batista, and Tianquan Lian. The impact of electric fields on processes at electrode interfaces.Chemical Reviews, 125:1604–1628, 2025
work page 2025
-
[11]
Ravishankar Sundararaman, William A. Goddard, and Tomas A. Arias. Grand canonical electronic density-functional theory: Algorithms and applications to electrochemistry.The Journal of Chemical Physics, 146(11):114104, 2017
work page 2017
-
[12]
Hörmann, Oliviero Andreussi, and Nicola Marzari
Nicolas G. Hörmann, Oliviero Andreussi, and Nicola Marzari. Grand canonical simulations of electrochemical interfaces in implicit solvation models.The Journal of Chemical Physics, 150:041730, 2019. doi: 10.1063/1.5054580. 18
-
[13]
Axel Groß. Grand-canonical approaches to understand structures and processes at electro- chemical interfaces from an atomistic perspective.Current Opinion in Electrochemistry, 27: 100684, 2021
work page 2021
-
[14]
Ravishankar Sundararaman, Derek Vigil-Fowler, and Kathleen Schwarz. Improving the accuracy of atomistic simulations of the electrochemical interface.Chemical Reviews, 122: 10651–10674, 2022
work page 2022
-
[15]
Ab initio simulations of water/metal interfaces.Chemical Reviews, 122:10746–10776, 2022
Axel Groß and Sung Sakong. Ab initio simulations of water/metal interfaces.Chemical Reviews, 122:10746–10776, 2022
work page 2022
-
[16]
Melander, Tongwei Wu, Timo Weckman, and Karoliina Honkala
Marko M. Melander, Tongwei Wu, Timo Weckman, and Karoliina Honkala. Constant inner potential DFT for modelling electrochemical systems under constant potential and bias. npj Computational Materials, 10:5, 2024
work page 2024
-
[17]
Ilja Siepmann and Michiel Sprik
J. Ilja Siepmann and Michiel Sprik. Influence of surface topology and electrostatic potential on water/electrode systems.The Journal of Chemical Physics, 102:511–524, 1995
work page 1995
-
[18]
GuillaumeJeanmairet, BenjaminRotenberg, andMathieuSalanne. Microscopicsimulations of electrochemical double-layer capacitors.Chemical Reviews, 122:10860–10898, 2022
work page 2022
-
[19]
Kim, Dong Hyun Kim, Junsic Cho, Seung-Jae Shin, Chang Hyuck Choi, and Hyungjun Kim
Minho M. Kim, Dong Hyun Kim, Junsic Cho, Seung-Jae Shin, Chang Hyuck Choi, and Hyungjun Kim. Electric double layer structure in concentrated aqueous solution.Nature Communications, 17:3645, 2026
work page 2026
-
[20]
Albert P. Bartók, Mike C. Payne, Risi Kondor, and Gábor Csányi. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons.Physical Review Letters, 104:136403, 2010
work page 2010
-
[21]
Alexander V. Shapeev. Moment tensor potentials: A class of systematically improvable interatomic potentials.Multiscale Modeling & Simulation, 14:1153–1173, 2016
work page 2016
-
[22]
Smith, Olexandr Isayev, and Adrian E
Justin S. Smith, Olexandr Isayev, and Adrian E. Roitberg. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost.Chemical Science, 8:3192–3203, 2017
work page 2017
-
[23]
Linfeng Zhang, Jiequn Han, Han Wang, Roberto Car, and Weinan E. Deep potential molecular dynamics: A scalable model with the accuracy of quantum mechanics.Physical Review Letters, 120:143001, 2018
work page 2018
-
[24]
Atomic cluster expansion for accurate and transferable interatomic potentials
Ralf Drautz. Atomic cluster expansion for accurate and transferable interatomic potentials. Physical Review B, 99:014104, 2019
work page 2019
-
[25]
Ilyes Batatia, Dávid Péter Kovács, Gregor N. C. Simm, Christoph Ortner, and Gábor Csányi. MACE: Higher order equivariant message passing neural networks for fast and accurate force fields. InAdvances in Neural Information Processing Systems (NeurIPS), volume 35, pages 11423–11436, 2022
work page 2022
-
[26]
Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, and Boris Kozinsky. E(3)-equivariant graph neu- ral networks for data-efficient and accurate interatomic potentials.Nature Communications, 13:2453, 2022
work page 2022
-
[27]
Owen, Mordechai Kornbluth, and Boris Kozinsky
Albert Musaelian, Simon Batzner, Anders Johansson, Lixin Sun, Cameron J. Owen, Mordechai Kornbluth, and Boris Kozinsky. Learning local equivariant representations for large-scale atomistic dynamics.Nature Communications, 14:579, 2023. 19
work page 2023
-
[28]
Finkler, Stefan Goedecker, and Jörg Behler
Tsz Wai Ko, Jonas A. Finkler, Stefan Goedecker, and Jörg Behler. General-purpose machine learning potentials capturing nonlocal charge transfer.Accounts of Chemical Research, 54: 808–817, 2021. doi: 10.1021/acs.accounts.0c00689
-
[29]
Dylan M. Anstine and Olexandr Isayev. Machine learning interatomic potentials and long- range physics.The Journal of Physical Chemistry A, 127:2417–2431, 2023
work page 2023
-
[30]
Nicolas Bergmann, Nicéphore Bonnet, Nicola Marzari, Karsten Reuter, and Nicolas G. Hörmann. Machine learning the energetics of electrified solid-liquid interfaces.Physical Review Letters, 135:146201, 2025
work page 2025
-
[31]
Ryosuke Jinnouchi and Saori Minami. Machine learning force fields in electrochemistry: From fundamentals to applications.ACS Nano, 19:22600–22644, 2025
work page 2025
-
[32]
Anthony K. Rappé and William A. Goddard. Charge equilibration for molecular dynamics simulations.The Journal of Physical Chemistry, 95:3358–3363, 1991
work page 1991
-
[33]
Alireza Ghasemi, Albert Hofstetter, Santanu Saha, and Stefan Goedecker
S. Alireza Ghasemi, Albert Hofstetter, Santanu Saha, and Stefan Goedecker. Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network.Physical Review B, 92:045131, 2015
work page 2015
-
[34]
Finkler, Stefan Goedecker, and Jörg Behler
Tsz Wai Ko, Jonas A. Finkler, Stefan Goedecker, and Jörg Behler. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer.Nature Communications, 12:398, 2021
work page 2021
-
[35]
Finkler, Philipp Misof, Tsz Wai Ko, Christoph Dellago, and Jörg Behler
Emir Kocer, Andreas Singraber, Jonas A. Finkler, Philipp Misof, Tsz Wai Ko, Christoph Dellago, and Jörg Behler. Iterative charge equilibration for fourth-generation high- dimensional neural network potentials.The Journal of Chemical Physics, 162:124106, 2025
work page 2025
-
[36]
Philip Fuchs, Maciej Sanocki, and Julija Zavadlav. Learning non-local molecular inter- actions via equivariant local representations and charge equilibration.npj Computational Materials, 11:287, 2025
work page 2025
-
[37]
Rongzhi Gao, ChiYung Yam, Jianjun Mao, Shuguang Chen, GuanHua Chen, and Ziyang Hu. A foundation machine learning potential with polarizable long-range interactions for materials modelling.Nature Communications, 16:10484, 2025
work page 2025
-
[38]
Yujing Wei, John L. Weber, James M. Stevenson, Zachary K. Goldsmith, Xiaowei Xie, Leif D. Jacobson, and Richard A. Friesner. An accurate charge-aware machine-learning interatomic potential for the reduction of Li-ion battery electrolytes in solution.Journal of Chemical Theory and Computation, 22(5):2515–2528, 2026
work page 2026
-
[39]
Nebgen, Sergei Tretiak, Joshua Finkelstein, Daniel P
Cheng-Han Li, Mehmet Cagri Kaymak, Maksim Kulichenko, Nicholas Lubbers, Ben- jamin T. Nebgen, Sergei Tretiak, Joshua Finkelstein, Daniel P. Tabor, and Anders M. N. Niklasson. Shadow molecular dynamics with a machine learned flexible charge potential. Journal of Chemical Theory and Computation, 21:3658–3675, 2025
work page 2025
-
[40]
Robert Stanton, Mehmet C. Kaymak, and Anders M. N. Niklasson. Shadow molecular dy- namics for a charge-potential equilibration model.Journal of Chemical Theory and Com- putation, 21:4779–4791, 2025
work page 2025
- [41]
-
[42]
Moritz Gubler, Jonas A. Finkler, Moritz R. Schäfer, Jörg Behler, and Stefan Goedecker. Accelerating fourth-generation machine learning potentials using quasi-linear scaling parti- cle mesh charge equilibration.Journal of Chemical Theory and Computation, 20:7264–7271, 2024
work page 2024
-
[43]
Active learning and explicit electrostatics enable accurate modeling of electrolytes
Olga Chalykh, Mikhail Polovinkin, Dmitry Korogod, Nikita Rybin, and Alexander Shapeev. Active learning and explicit electrostatics enable accurate modeling of electrolytes. arXiv:2510.03479, 2025
-
[44]
Martin Vondrák, Karsten Reuter, and Johannes T. Margraf. Pushing charge equilibration- based machine learning potentials to their limits.npj Computational Materials, 11:288, 2025
work page 2025
-
[45]
G. Lee Warren, Joseph E. Davis, and Sandeep Patel. Origin and control of superlinear polarizability scaling in chemical potential equalization methods.The Journal of Chemical Physics, 128:144110, 2008
work page 2008
-
[46]
Razvan A. Nistor, Jeliazko G. Polihronov, Martin H. Müser, and Nicholas J. Mosey. A generalization of the charge equilibration method for nonmetallic materials.The Journal of Chemical Physics, 125:094108, 2006
work page 2006
-
[47]
Razvan A. Nistor and Martin H. Müser. Dielectric properties of solids in the regular and split-charge equilibration formalisms and the point-dipole limit.Physical Review B, 79: 104303, 2009
work page 2009
-
[48]
Toon Verstraelen, Veronique Van Speybroeck, and Michel Waroquier. The electronega- tivity equalization method and the split charge equilibration applied to organic systems: Parametrization, validation, and comparison.The Journal of Chemical Physics, 131:044127, 2009
work page 2009
-
[49]
Ayers, Dimitri Van Neck, and Michel Waroquier
Toon Verstraelen, Patrick Bultinck, Veronique Van Speybroeck, Paul W. Ayers, Dimitri Van Neck, and Michel Waroquier. The significance of parameters in charge equilibration models.Journal of Chemical Theory and Computation, 7:1750–1764, 2011
work page 2011
-
[50]
Ayers, Veronique Van Speybroeck, and Michel Waroquier
Toon Verstraelen, Paul W. Ayers, Veronique Van Speybroeck, and Michel Waroquier. ACKS2: Atom-condensed Kohn–Sham DFT approximated to second order.The Journal of Chemical Physics, 138:074108, 2013
work page 2013
-
[51]
Benjamin Nebgen, Nicholas Lubbers, Justin S. Smith, Andrew E. Sifain, Andrey Lokhov, Olexandr Isayev, Adrian E. Roitberg, Kipton Barros, and Sergei Tretiak. Transferable dynamic molecular charge assignment using deep neural networks.Journal of Chemical Theory and Computation, 14:4687–4698, 2018
work page 2018
-
[52]
Anstine, Roman Zubatyuk, and Olexandr Isayev
Dylan M. Anstine, Roman Zubatyuk, and Olexandr Isayev. AIMNet2: A neural network potential to meet your neutral, charged, organic, and elemental-organic needs.Chemical Science, 16:10228–10244, 2025
work page 2025
-
[53]
King, Dongjin Kim, Peichen Zhong, and Bingqing Cheng
Daniel S. King, Dongjin Kim, Peichen Zhong, and Bingqing Cheng. Machine learning of charges and long-range interactions from energies and forces.Nature Communications, 16: 8763, 2025
work page 2025
-
[54]
Ruoyu Wang, Shaoheng Fang, Qixing Huang, and Yuanyue Liu. Constant-potential ma- chine learning force field for the electrochemical interface.Journal of Chemical Theory and Computation, 21:7628–7635, 2025. 21
work page 2025
-
[55]
Steven W. Rick, Steven J. Stuart, and B. J. Berne. Dynamical fluctuating charge force fields: Application to liquid water.The Journal of Chemical Physics, 101(7):6141–6156, 1994
work page 1994
-
[56]
Riccardo Chelli and Piero Procacci. A transferable polarizable electrostatic force field for molecular mechanics based on the chemical potential equalization principle.The Journal of Chemical Physics, 117(20):9175–9189, 2002
work page 2002
-
[57]
Nicholas Lubbers, Justin S. Smith, and Kipton Barros. Hierarchical modeling of molecular energies using a deep neural network.The Journal of Chemical Physics, 148:241715, 2018
work page 2018
-
[58]
Nicholas Lubbers et al. hippynn: A modular library for atomistic machine learning with PyTorch.https://github.com/lanl/hippynn, 2023
work page 2023
-
[59]
Thomas A. Manz and Nidia Gabaldon Limas. Introducing DDEC6 atomic population analysis: Part 1. charge partitioning theory and methodology.RSC Advances, 6:47771– 47801, 2016
work page 2016
-
[60]
Nongnuch Artrith, Tobias Morawietz, and Jörg Behler. High-dimensional neural-network potentials for multicomponent systems: Applications to zinc oxide.Physical Review B, 83: 153101, 2011
work page 2011
-
[61]
Oliver T. Unke and Markus Meuwly. PhysNet: A neural network for predicting energies, forces, dipole moments, and partial charges.Journal of Chemical Theory and Computation, 15:3678–3693, 2019
work page 2019
-
[62]
Peter G. Doyle and J. Laurie Snell.Random Walks and Electric Networks. Mathematical Association of America, 1984
work page 1984
-
[63]
Todd R. Gingrich and Mark Wilson. On the Ewald summation of Gaussian charges for the simulation of metallic surfaces.Chemical Physics Letters, 500(1–3):178–183, 2010. doi: 10.1016/j.cplett.2010.10.010
-
[64]
Satomichi Nishihara and Minoru Otani. Hybrid solvation models for bulk, interface, and membrane: Referenceinteractionsitemethodscoupledwithdensityfunctionaltheory.Phys- ical Review B, 96:115429, 2017
work page 2017
-
[65]
Georg Kresse and Jürgen Furthmüller. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set.Physical Review B, 54:11169–11186, 1996
work page 1996
-
[66]
Perdew, Kieron Burke, and Matthias Ernzerhof
John P. Perdew, Kieron Burke, and Matthias Ernzerhof. Generalized gradient approxima- tion made simple.Physical Review Letters, 77(18):3865–3868, 1996
work page 1996
-
[67]
Stefan Grimme, Jens Antony, Stephan Ehrlich, and Helge Krieg. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu.Journal of Chemical Physics, 132(15):154104, 2010
work page 2010
-
[68]
Stefan Grimme, Stephan Ehrlich, and Lars Goerigk. Effect of the damping function in dispersion corrected density functional theory.Journal of Computational Chemistry, 32(7): 1456–1465, 2011
work page 2011
-
[69]
Introducingddec6atomicpopulationanalysis: part 4
NidiaGabaldonLimasandThomasA.Manz. Introducingddec6atomicpopulationanalysis: part 4. efficient parallel computation of net atomic charges, atomic spin moments, bond orders, and more.RSC Advances, 8:2678–2707, 2018. 22
work page 2018
-
[70]
Maksim Kulichenko, Benjamin Nebgen, Nicholas Lubbers, Justin S. Smith, Kipton Barros, Alice E. A. Allen, Adela Habib, Emily Shinkle, Nikita Fedik, Ying Wai Li, Richard A. Messerly, and Sergei Tretiak. Data generation for machine learning interatomic potentials and beyond.Chemical Reviews, 124:13681–13714, 2024
work page 2024
-
[71]
Lauri Himanen, Marc O. J. Jäger, Eiaki V. Morooka, Filippo Federici Canova, Yashasvi S. Ranawat, David Z. Gao, Patrick Rinke, and Adam S. Foster. DScribe: Library of descriptors for machine learning in materials science.Computer Physics Communications, 247:106949, 2020
work page 2020
-
[72]
Engel, Jörg Behler, Christoph Dellago, and Michele Ceriotti
Bingqing Cheng, Edgar A. Engel, Jörg Behler, Christoph Dellago, and Michele Ceriotti. Ab initio thermodynamics of liquid and solid water.Proceedings of the National Academy of Sciences, 116(4):1110–1115, 2019
work page 2019
-
[73]
Justin S. Smith, Benjamin Nebgen, Nithin Mathew, Jie Chen, Nicholas Lubbers, Leonid Burakovsky, Sergei Tretiak, Hai Ah Nam, Timothy Germann, Saryu Fensin, and Kipton Barros. Automated discovery of a robust interatomic potential for aluminum.Nature Communications, 12:1257, 2021
work page 2021
-
[74]
Ludwig J. V. Ahrens-Iwers, Mathijs Janssen, Shern R. Tee, and Robert H. Meißner. ELEC- TRODE: An electrochemistry package for atomistic simulations.The Journal of Chemical Physics, 157:084801, 2022
work page 2022
-
[75]
Adri C. T. van Duin, Siddharth Dasgupta, Francois Lorant, and William A. Goddard. ReaxFF: A reactive force field for hydrocarbons.The Journal of Physical Chemistry A, 105:9396–9409, 2001
work page 2001
-
[76]
Nicolas Onofrio and Alejandro Strachan. Voltage equilibration for reactive atomistic simu- lations of electrochemical processes.The Journal of Chemical Physics, 143:054109, 2015. 23
work page 2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.