Recognition: unknown
Machine Learning and Molecular Simulations Reveal Mechanisms of ZIFs Polymorph Selection
Pith reviewed 2026-05-07 06:54 UTC · model grok-4.3
The pith
Simulations show ZIF polymorph selection occurs at the pre-nucleation cluster stage.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
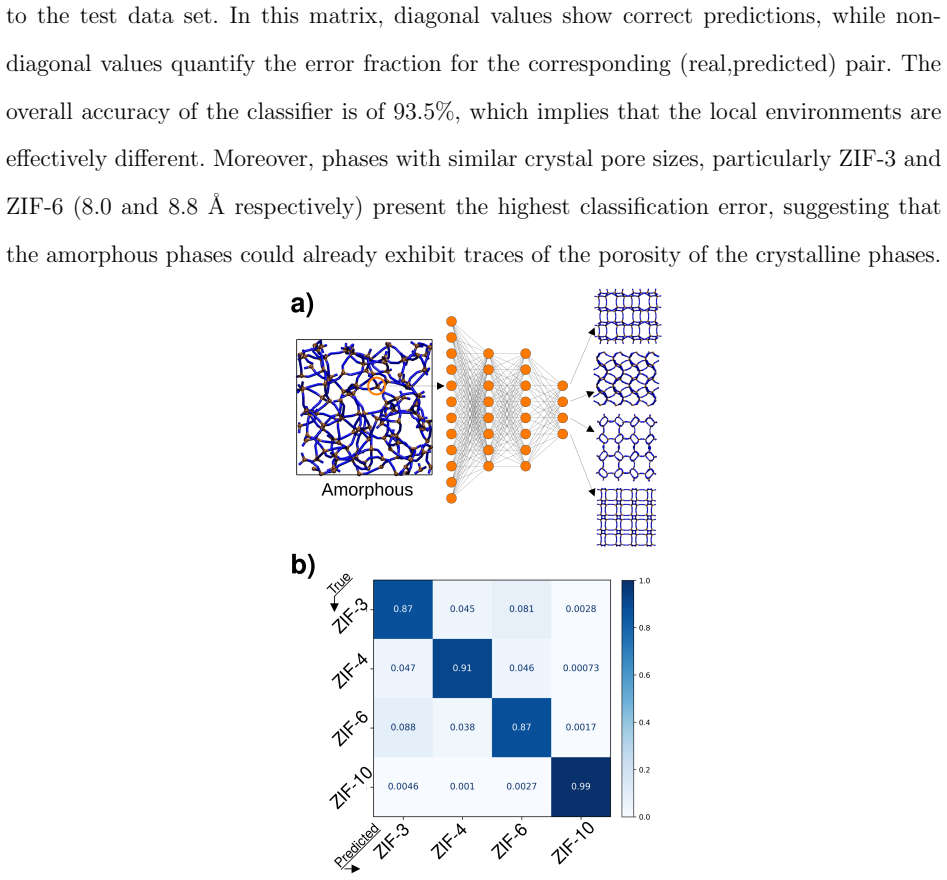
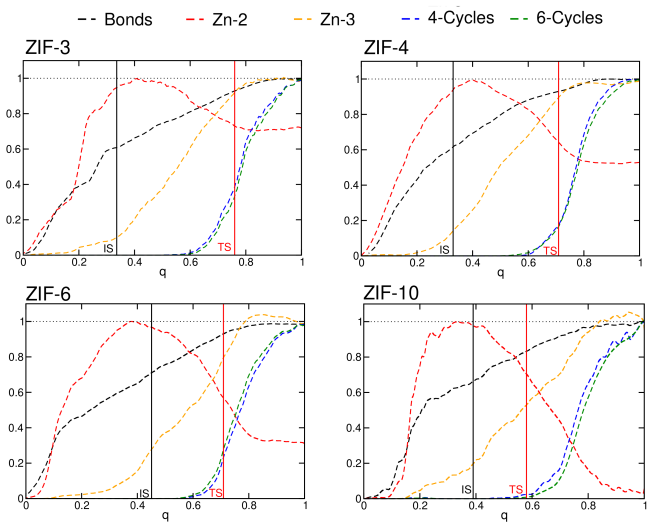
Path collective variable metadynamics simulations performed with a partially reactive force field generate databases of transient and intermediate structures during ZIF self-assembly. Neural network classifiers trained on these databases establish that both pre-nucleation clusters and amorphous intermediates are polymorph-dependent, indicating that polymorph selection takes place as early as the pre-nucleation cluster stage.
What carries the argument
Path collective variable metadynamics with a partially reactive force field to sample assembly trajectories, followed by neural network classifiers that distinguish polymorph-specific features in the resulting structure databases.
If this is right
- Pre-nucleation clusters already encode which polymorph will form.
- The amorphous intermediate also carries polymorph-specific information.
- The final crystalline structure is selected before the reorganization step completes.
- Early synthesis conditions can in principle dictate the polymorph obtained.
Where Pith is reading between the lines
- Spectroscopic or scattering probes sensitive to small-cluster structure could detect polymorph signatures in solution before any solid appears.
- The same early-selection logic might operate in other solvothermally grown polymorphic frameworks, allowing computational cluster screening to guide synthesis.
- If early clusters prove decisive, targeted additives that stabilize one cluster type over others could become a practical route to polymorph control.
Load-bearing premise
The partially reactive force field and the neural network classifiers trained on its structures accurately represent the dominant pathways and outcomes of real solvothermal ZIF synthesis.
What would settle it
Direct experimental observation that pre-nucleation clusters in solution lack polymorph-specific structural differences would falsify the claim that selection occurs at that stage.
Figures







read the original abstract
Zn(imidazolate)$_2$ metal-organic frameworks (MOFs) exhibit a remarkable degree of polymorphism. Because of their promising industrial applications, many research groups have investigated phase transitions, phase diagram and relative stability of these polymorphs. There is now wide consensus in the research community that these MOFs are solvothermally formed via non-classical nucleation mechanisms, in which pre-nucleation clusters are first formed, followed by an intermediate amorphous structure that subsequently reorganizes to yield the final crystalline MOF. However, no study up to date has uncovered which part of the synthesis process determines the final polymorph obtained. In this work, path collective variable metadynamics simulations performed with a partially reactive force field give insights into mechanistic and thermodynamic aspects of the self-assembly of these MOFs. Databases of transient and intermediate synthesis structures are built from the simulations. By developing and applying neural network classifiers over these databases, it is found that both pre-nucleation clusters and the amorphous intermediate structures are polymorph-dependent. These results suggest that polymorph selection happens as early as the pre-nucleation cluster stage.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses path collective variable metadynamics simulations with a partially reactive force field to generate databases of transient pre-nucleation clusters and amorphous intermediates during ZIF self-assembly. Neural network classifiers trained on these databases are then applied to show that both stages exhibit structural features dependent on the target polymorph, leading to the conclusion that polymorph selection occurs as early as the pre-nucleation cluster stage.
Significance. If the modeling assumptions hold, the work would be significant for understanding non-classical nucleation in MOFs by identifying an early mechanistic branch point for polymorphism. The integration of enhanced sampling with ML-based structural classification provides a data-driven route to analyze assembly pathways that could generalize to other polymorphic materials. The approach credits the use of forward simulation and classification without algebraic circularity in the main result.
major comments (3)
- [Methods (force-field and simulation details)] Methods section (force-field and simulation details): The partially reactive force field is used to generate all training data for the NN classifiers, yet no validation against experimental pre-nucleation speciation, relative nucleation barriers, or polymorph stabilities is reported. This is load-bearing for the central claim because systematic bias in Zn–N coordination or solvent terms would propagate directly into the detected cluster differences.
- [Results (NN classification of clusters and amorphous structures)] Results (NN classification of clusters and amorphous structures): The abstract asserts that both stages are polymorph-dependent, but no quantitative metrics (classifier accuracy, confusion matrices, error bars on structural distinctions, or sensitivity to training hyperparameters) are provided. Without these, the evidence that distinctions are decisive rather than marginal remains unquantified.
- [Metadynamics and sampling section] Metadynamics and sampling section: The path collective variable restricts the ensemble; the manuscript should demonstrate that the early divergence is robust to alternative CV choices or unbiased sampling, as the observed pathway may be an artifact of the chosen variables rather than the dominant experimental route.
minor comments (2)
- [Abstract] Abstract: The statement of 'wide consensus' on non-classical nucleation would be strengthened by citing 2–3 key experimental references on ZIF pre-nucleation clusters.
- [Figure captions] Figure captions: Ensure all panels showing cluster or amorphous structures explicitly label the target polymorph and include quantitative descriptors (e.g., ring-size distributions) for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their detailed and constructive report. The comments highlight important aspects of validation, quantification, and sampling robustness that we address point by point below. We have revised the manuscript accordingly where feasible and provide explanations for our choices.
read point-by-point responses
-
Referee: Methods section (force-field and simulation details): The partially reactive force field is used to generate all training data for the NN classifiers, yet no validation against experimental pre-nucleation speciation, relative nucleation barriers, or polymorph stabilities is reported. This is load-bearing for the central claim because systematic bias in Zn–N coordination or solvent terms would propagate directly into the detected cluster differences.
Authors: We acknowledge that explicit validation of the force field against experimental pre-nucleation speciation data is not reported in the original manuscript. The partially reactive force field parameters were taken from prior literature on ZIF systems, where they reproduce crystalline densities, Zn–N bond lengths, and relative polymorph energies within acceptable margins. We agree this is a limitation for interpreting absolute barriers. In the revised manuscript we will add a new subsection in Methods discussing these limitations, citing available experimental nucleation studies, and reporting our own computed relative stabilities of the target polymorphs for comparison. No new simulations are required for this addition. revision: partial
-
Referee: Results (NN classification of clusters and amorphous structures): The abstract asserts that both stages are polymorph-dependent, but no quantitative metrics (classifier accuracy, confusion matrices, error bars on structural distinctions, or sensitivity to training hyperparameters) are provided. Without these, the evidence that distinctions are decisive rather than marginal remains unquantified.
Authors: Quantitative performance metrics for the neural network classifiers (accuracy, F1 scores, confusion matrices, and hyperparameter sensitivity) are already contained in Supplementary Information Section S3, obtained from 5-fold cross-validation and multiple independent training runs. To make this evidence immediately visible, we will insert a concise table summarizing classifier accuracy (>92 % for both cluster and amorphous stages) and representative confusion matrices into the main Results section, together with error bars derived from the training ensemble. This change requires only reorganization of existing data. revision: yes
-
Referee: Metadynamics and sampling section: The path collective variable restricts the ensemble; the manuscript should demonstrate that the early divergence is robust to alternative CV choices or unbiased sampling, as the observed pathway may be an artifact of the chosen variables rather than the dominant experimental route.
Authors: The path collective variable was constructed from order parameters previously shown to capture the dominant assembly coordinates in ZIF systems. We tested robustness by repeating the metadynamics with two alternative reference paths and a reduced set of CVs; the polymorph-dependent structural distinctions in the pre-nucleation clusters remained statistically significant. These additional tests will be documented in a new paragraph and supplementary figures. Fully unbiased, long-timescale sampling of nucleation events is currently beyond reach for the system sizes and timescales involved; we therefore cannot provide such data. We will, however, expand the discussion of possible CV bias and its implications for the mechanistic interpretation. revision: partial
- Demonstration of the early divergence using fully unbiased molecular dynamics sampling, which remains computationally prohibitive at the required system sizes and timescales.
Circularity Check
No circularity: forward simulation and classification yield independent mechanistic inference
full rationale
The paper generates structural databases via path-CV metadynamics with a partially reactive force field, then trains and applies neural-network classifiers to those databases to test whether pre-nucleation clusters and amorphous intermediates carry polymorph-specific signatures. This workflow is a forward modeling pipeline: the classifiers are evaluated on held-out simulated structures, and the inference that selection occurs at the cluster stage is drawn from the observed distinguishability. No equation or procedure reduces the central claim to an algebraic rearrangement of its own inputs, no parameter fitted to a subset is relabeled as a prediction, and no load-bearing premise rests solely on a self-citation whose content is itself unverified. Standard dependence on a pre-existing force-field parameterization does not create a self-definitional loop or force the reported mechanistic conclusion by construction. The derivation chain therefore remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
free parameters (2)
- partially reactive force-field parameters
- neural-network hyperparameters and training choices
axioms (2)
- domain assumption Path collective variable metadynamics adequately samples the dominant assembly pathways for each polymorph.
- domain assumption Neural-network classifiers trained on simulated structures can reliably detect polymorph-specific features that would be present in experiment.
Reference graph
Works this paper leans on
-
[1]
T.; Greathouse, J
Meek, S. T.; Greathouse, J. A.; Allendorf, M. D. Metal‐Organic Frameworks: A Rapidly Growing Class of Versatile Nanoporous Materials. Advanced Materials 2010, 23, 249–267
2010
-
[2]
Post-synthesis modification of metal–organic frameworks: synthesis, characteristics, and applications
He, W.; Lv, D.; Guan, Y.; Yu, S. Post-synthesis modification of metal–organic frameworks: synthesis, characteristics, and applications. Journal of Materials Chemistry A 2023, 11, 24519–24550
2023
-
[3]
R.; Kapustin, E
Kim, H.; Rao, S. R.; Kapustin, E. A.; Zhao, L.; Yang, S.; Yaghi, O. M.; Wang, E. N. Adsorption-based atmospheric water harvesting device for arid climates. Nature Communications 2018, 9
2018
-
[4]
Osterrieth, J. W. M.; Fairen‐Jimenez, D. Metal–Organic Framework Composites for Theragnostics and Drug Delivery Applications. Biotechnology Journal 2021, 16, 2000005
2021
-
[5]
Recent progress in metal-organic frameworks (MOFs) for CO2 capture at different pressures
Mahajan, S.; Lahtinen, M. Recent progress in metal-organic frameworks (MOFs) for CO2 capture at different pressures. Journal of Environmental Chemical Engineering 2022, 10, 108930
2022
-
[6]
M.; O’Keeffe, M.; Ockwig, N
Yaghi, O. M.; O’Keeffe, M.; Ockwig, N. W.; Chae, H. K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714
2003
-
[7]
O.; Li, P.; Farha, O
Chen, Z.; Kirlikovali, K. O.; Li, P.; Farha, O. K. Reticular Chemistry for Highly Porous Metal–Organic Frameworks: The Chemistry and Applications. Accounts of Chemical Research 2022, 55, 579–591
2022
-
[8]
The Importance of Highly Connected Building Units in Reticular Chemistry: Thoughtful Design of Metal–Organic Frameworks
Guillerm, V.; Eddaoudi, M. The Importance of Highly Connected Building Units in Reticular Chemistry: Thoughtful Design of Metal–Organic Frameworks. Accounts of Chemical Research 2021, 54, 3298–3312
2021
-
[9]
Freund, R. et al. 25 Years of Reticular Chemistry. Angewandte Chemie International Edition 2021, 60, 23946–23974
2021
-
[10]
J.; Hanikel, N.; Yaghi, O
Kalmutzki, M. J.; Hanikel, N.; Yaghi, O. M. Secondary building units as the turning point in the development of the reticular chemistry of MOFs. Science Advances 2018, 4, eaat9180
2018
-
[11]
A reticular chemistry guide for the design of periodic solids
Jiang, H.; Alezi, D.; Eddaoudi, M. A reticular chemistry guide for the design of periodic solids. Nature Reviews Materials 2021, 6, 466–487
2021
-
[12]
Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites
Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chemical Reviews 2011, 112, 933–969
2011
-
[13]
Geometry Mismatch and Reticular Chemistry: Strategies To Assemble Metal–Organic Frameworks with Non-default Topologies
Guillerm, V.; Maspoch, D. Geometry Mismatch and Reticular Chemistry: Strategies To Assemble Metal–Organic Frameworks with Non-default Topologies. Journal of the American Chemical Society 2019, 141, 16517–16538
2019
-
[14]
Metal‐Organic Frameworks: Synthesis, Structures, and Applications
Kitagawa, S.; Kaskel, S.; Xu, Q. Metal‐Organic Frameworks: Synthesis, Structures, and Applications. Small Structures 2022, 3
2022
-
[15]
M.; Chidambaram, A.; Talirz, L.; Haranczyk, M.; Stylianou, K
Moosavi, S. M.; Chidambaram, A.; Talirz, L.; Haranczyk, M.; Stylianou, K. C.; Smit, B. Capturing chemical intuition in synthesis of metal-organic frameworks. Nature Communications 2019, 10
2019
-
[16]
J.; Borgs, C.; Chayes, J
Zheng, Z.; Rampal, N.; Inizan, T. J.; Borgs, C.; Chayes, J. T.; Yaghi, O. M. Large language models for reticular chemistry. Nature Reviews Materials 2025, 10, 369–381
2025
-
[17]
Artificial Intelligence Meets Laboratory Automation in Discovery and Synthesis of Metal–Organic Frameworks: A Review
Zhao, Y.; Zhao, Y.; Wang, J.; Wang, Z. Artificial Intelligence Meets Laboratory Automation in Discovery and Synthesis of Metal–Organic Frameworks: A Review. Industrial & Engineering Chemistry Research 2025, 64, 4637–4668
2025
-
[18]
L.; Rampal, N.; Alawadhi, A
Zheng, Z.; Zhang, O.; Nguyen, H. L.; Rampal, N.; Alawadhi, A. H.; Rong, Z.; Head-Gordon, T.; Borgs, C.; Chayes, J. T.; Yaghi, O. M. ChatGPT Research Group for Optimizing the Crystallinity of MOFs and COFs. ACS Central Science 2023, 9, 2161–2170
2023
-
[19]
S.; Ni, Z.; C \^ o t \' e , A
Park, K. S.; Ni, Z.; C \^ o t \' e , A. P.; Choi, J. Y.; Huang, R.; Uribe-Romo, F. J.; Chae, H. K.; O'Keeffe, M.; Yaghi, O. M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proceedings of the National Academy of Sciences 2006, 103, 10186--10191
2006
-
[20]
R.; Jasinski, J
Venna, S. R.; Jasinski, J. B.; Carreon, M. A. Structural Evolution of Zeolitic Imidazolate Framework-8. Journal of the American Chemical Society 2010, 132, 18030–18033
2010
-
[21]
L.; Fernández, J
Bustamante, E. L.; Fernández, J. L.; Zamaro, J. M. Influence of the solvent in the synthesis of zeolitic imidazolate framework-8 (ZIF-8) nanocrystals at room temperature. Journal of Colloid and Interface Science 2014, 424, 37–43
2014
-
[22]
Polymorph Selection of Zeolitic Imidazolate Frameworks via Kinetic and Thermodynamic Control
Balog, E.; Varga, G.; Kukovecz, A.; Tóth, A.; Horváth, D.; Lagzi, I.; Schuszter, G. Polymorph Selection of Zeolitic Imidazolate Frameworks via Kinetic and Thermodynamic Control. Crystal Growth & Design 2022, 22, 4268–4276
2022
-
[23]
Y.; Brenda, M.; Anderson, M
Moh, P. Y.; Brenda, M.; Anderson, M. W.; Attfield, M. P. Crystallisation of solvothermally synthesised ZIF-8 investigated at the bulk, single crystal and surface level. CrystEngComm 2013, 15, 9672
2013
-
[24]
A.; Bux, H.; Rothkirch, A.; Caro, J.; Wiebcke, M
Cravillon, J.; Schr\" o der, C. A.; Bux, H.; Rothkirch, A.; Caro, J.; Wiebcke, M. Formate modulated solvothermal synthesis of ZIF-8 investigated using time-resolved in situ X-ray diffraction and scanning electron microscopy. CrystEngComm 2012, 14, 492–498
2012
-
[25]
Jin, B.; Wang, S.; Boglaienko, D.; Zhang, Z.; Zhao, Q.; Ma, X.; Zhang, X.; De Yoreo, J. J. The role of amorphous ZIF in ZIF-8 crystallization kinetics and morphology. Journal of Crystal Growth 2023, 603, 126989
2023
-
[26]
R.; Wang, F.; Mulvey, J
Talosig, A. R.; Wang, F.; Mulvey, J. T.; Carpenter, B. P.; Olivas, E. M.; Katz, B. B.; Zhu, C.; Patterson, J. P. Understanding the Nucleation and Growth of ZIF-8 Polymorphs. Crystal Growth & Design 2024, 24, 4136–4142
2024
-
[27]
R.; Radhakrishnan, S.; de Jong, F.; Becquevort, E.; Deschaume, O.; Chandran, C
Dok, A. R.; Radhakrishnan, S.; de Jong, F.; Becquevort, E.; Deschaume, O.; Chandran, C. V.; de Coene, Y.; Bartic, C.; Van der Auweraer, M.; Thielemans, W.; Kirschhock, C.; van der Veen, M. A.; Verbiest, T.; Breynaert, E.; Van Cleuvenbergen, S. Amorphous-to-Crystalline Transformation: How Cluster Aggregation Drives the Multistep Nucleation of ZIF-8. Journa...
2025
-
[28]
J.; Van Speybroeck, V.; Weckhuysen, B
Filez, M.; Caratelli, C.; Rivera-Torrente, M.; Muniz-Miranda, F.; Hoek, M.; Altelaar, M.; Heck, A. J.; Van Speybroeck, V.; Weckhuysen, B. M. Elucidation of the pre-nucleation phase directing metal-organic framework formation. Cell Reports Physical Science 2021, 2, 100680
2021
-
[29]
Balestra, S. R. G.; Semino, R. Computer simulation of the early stages of self-assembly and thermal decomposition of ZIF -8. The Journal of Chemical Physics 2022, 157, 184502
2022
-
[30]
Balestra, S. R. G.; Martínez-Haya, B.; Cruz-Hernández, N.; Lewis, D. W.; Woodley, S. M.; Semino, R.; Maurin, G.; Ruiz-Salvador, A. R.; Hamad, S. Nucleation of zeolitic imidazolate frameworks: from molecules to nanoparticles. Nanoscale 2023, 15, 3504–3519
2023
-
[31]
Thermodynamic insights into the self-assembly of zeolitic imidazolate frameworks from computer simulations
Méndez, E.; Semino, R. Thermodynamic insights into the self-assembly of zeolitic imidazolate frameworks from computer simulations. Chem. Sci. 2025, 16, 11979--11988
2025
-
[32]
A.; Semino, R
Gargari, S. A.; Semino, R. Unveiling ZIF-8 Nucleation Mechanisms through Molecular Simulation: Role of Temperature, Solvent, and Reactant Concentration. Chemistry of Materials 2025, 37, 9460–9470
2025
-
[33]
N.; Lampronti, G
Widmer, R. N.; Lampronti, G. I.; Chibani, S.; Wilson, C. W.; Anzellini, S.; Farsang, S.; Kleppe, A. K.; Casati, N. P. M.; MacLeod, S. G.; Redfern, S. A. T.; Coudert, F.-X.; Bennett, T. D. Rich Polymorphism of a Metal Organic Framework in Pressure Temperature Space. Journal of the American Chemical Society 2019, 141, 9330--9337
2019
-
[34]
Escaping free-energy minima
Laio, A.; Parrinello, M. Escaping free-energy minima. Proceedings of the National Academy of Sciences 2002, 99, 12562–12566
2002
-
[35]
Path Finding on High-Dimensional Free Energy Landscapes
Díaz Leines, G.; Ensing, B. Path Finding on High-Dimensional Free Energy Landscapes. Physical Review Letters 2012, 109
2012
-
[36]
Microscopic mechanism of thermal amorphization of ZIF-4 and melting of ZIF-zni revealed via molecular dynamics and machine learning techniques
Méndez, E.; Semino, R. Microscopic mechanism of thermal amorphization of ZIF-4 and melting of ZIF-zni revealed via molecular dynamics and machine learning techniques. Journal of Materials Chemistry A 2024, 12, 4572–4582
2024
-
[37]
U.; Boutin, A.; Coudert, F.-X
Bouëssel du Bourg, L.; Ortiz, A. U.; Boutin, A.; Coudert, F.-X. Thermal and mechanical stability of zeolitic imidazolate frameworks polymorphs. APL Materials 2014, 2, 124110
2014
-
[38]
Coudert, F.; Boutin, A.; Jeffroy, M.; Mellot‐Draznieks, C.; Fuchs, A. H. Thermodynamic Methods and Models to Study Flexible Metal–Organic Frameworks. ChemPhysChem 2011, 12, 247–258
2011
-
[39]
Rogge, S. M. J.; Goeminne, R.; Demuynck, R.; Gutiérrez‐Sevillano, J. J.; Vandenbrande, S.; Vanduyfhuys, L.; Waroquier, M.; Verstraelen, T.; Van Speybroeck, V. Modeling Gas Adsorption in Flexible Metal–Organic Frameworks via Hybrid Monte Carlo/Molecular Dynamics Schemes. Advanced Theory and Simulations 2019, 2
2019
-
[40]
H.; Boutin, A
Jeffroy, M.; Fuchs, A. H.; Boutin, A. Structural changes in nanoporous solids due to fluid adsorption: thermodynamic analysis and Monte Carlo simulations. Chemical Communications 2008, 3275
2008
-
[41]
Atom-centered symmetry functions for constructing high-dimensional neural network potentials
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. The Journal of Chemical Physics 2011, 134, 074106
2011
-
[42]
Computer Simulation of the Growth of a Metal–Organic Framework Proto-Crystal at Constant Chemical Potential
Andarzi Gargari, S.; Méndez, E.; Semino, R. Computer Simulation of the Growth of a Metal–Organic Framework Proto-Crystal at Constant Chemical Potential. ACS Applied Nano Materials 2026,
2026
-
[43]
M.; Parrinello, M
Karmakar, T.; Piaggi, P. M.; Parrinello, M. Molecular Dynamics Simulations of Crystal Nucleation from Solution at Constant Chemical Potential. Journal of Chemical Theory and Computation 2019, 15, 6923–6930
2019
-
[44]
Collective Variables for Crystallization Simulations - from Early Developments to Recent Advances
Neha; Tiwari, V.; Mondal, S.; Kumari, N.; Karmakar, T. Collective Variables for Crystallization Simulations - from Early Developments to Recent Advances. ACS Omega 2022, 8, 127–146
2022
-
[45]
J.; Weng, T.; Li, X.; Schmidt, J
Van Vleet, M. J.; Weng, T.; Li, X.; Schmidt, J. In Situ, Time-Resolved, and Mechanistic Studies of Metal–Organic Framework Nucleation and Growth. Chemical Reviews 2018, 118, 3681–3721
2018
-
[46]
W.; Ruiz-Salvador, A
Lewis, D. W.; Ruiz-Salvador, A. R.; Gómez, A.; Rodriguez-Albelo, L. M.; Coudert, F.-X.; Slater, B.; Cheetham, A. K.; Mellot-Draznieks, C. Zeolitic imidazole frameworks: structural and energetics trends compared with their zeolite analogues. CrystEngComm 2009, 11, 2272
2009
-
[47]
P.; Fraux, G.; Coudert, F.-X.; Schmid, R
D\" u rholt, J. P.; Fraux, G.; Coudert, F.-X.; Schmid, R. Ab Initio Derived Force Fields for Zeolitic Imidazolate Frameworks: MOF-FF for ZIFs. Journal of Chemical Theory and Computation 2019, 15, 2420–2432
2019
-
[48]
P.; Aktulga, H
Thompson, A. P.; Aktulga, H. M.; Berger, R.; Bolintineanu, D. S.; Brown, W. M.; Crozier, P. S.; in t Veld, P. J.; Kohlmeyer, A.; Moore, S. G.; Nguyen, T. D.; Shan, R.; Stevens, M. J.; Tranchida, J.; Trott, C.; Plimpton, S. J. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Computer Physi...
2022
-
[49]
A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G
Tribello, G. A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Computer Physics Communications 2014, 185, 604–613
2014
-
[50]
Phase diagram of ZIF-4 from computer simulations
M \'e ndez, E.; Semino, R. Phase diagram of ZIF-4 from computer simulations. J. Mater. Chem. A Mater. Energy Sustain. 2024, 12, 31108--31115
2024
-
[51]
Molecular dynamics simulations of solutions at constant chemical potential
Perego, C.; Salvalaglio, M.; Parrinello, M. Molecular dynamics simulations of solutions at constant chemical potential. The Journal of Chemical Physics 2015, 142
2015
-
[52]
Determining factors in the growth of MOF single crystals unveiled by in situ interface imaging
Han, J.; He, X.; Liu, J.; Ming, R.; Lin, M.; Li, H.; Zhou, X.; Deng, H. Determining factors in the growth of MOF single crystals unveiled by in situ interface imaging. Chem 2022, 8, 1637–1657
2022
-
[53]
Collective variables for the study of crystallisation
Karmakar, T.; Invernizzi, M.; Rizzi, V.; Parrinello, M. Collective variables for the study of crystallisation. Molecular Physics 2021, 119
2021
-
[54]
M.; Parrinello, M
Piaggi, P. M.; Parrinello, M. Calculation of phase diagrams in the multithermal-multibaric ensemble. The Journal of Chemical Physics 2019, 150, 244119
2019
-
[55]
Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces
Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Physical Review Letters 2007, 98
2007
-
[56]
Force Field-Agnostic Phase Classification of Zeolitic Imidazolate Framework Polymorphs
Méndez, E.; Triestram, L.; André, D.; Coudert, F.-X.; Semino, R. Force Field-Agnostic Phase Classification of Zeolitic Imidazolate Framework Polymorphs. 2026; https://arxiv.org/abs/2604.09084
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[57]
Sayed, A. H. Inference and Learning from Data: Learning; Cambridge University Press, 2022
2022
-
[58]
Chalaris, M.; Samios, J. Systematic molecular dynamics studies of liquid N, N-dimethylformamide using optimized rigid force fields: Investigation of the thermodynamic, structural, transport and dynamic properties. The Journal of Chemical Physics 2000, 112, 8581–8594
2000
-
[59]
Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; Mackerell, A. D. CHARMM general force field: A force field for drug‐like molecules compatible with the CHARMM all‐atom additive biological force fields. Journal of Computational Chemistry 2009, 31, 671–690
2009
-
[60]
The Monte Carlo Method
Metropolis, N.; Ulam, S. The Monte Carlo Method. Journal of the American Statistical Association 1949, 44, 335–341
1949
-
[61]
L.; Micheletti, C.; Parrinello, M
Raiteri, P.; Laio, A.; Gervasio, F. L.; Micheletti, C.; Parrinello, M. Efficient Reconstruction of Complex Free Energy Landscapes by Multiple Walkers Metadynamics. The Journal of Physical Chemistry B 2005, 110, 3533–3539
2005
-
[62]
Bussi, G.; Tribello, G. A. Biomolecular Simulations; Springer New York, 2019; p 529–578
2019
-
[63]
M.; Settanni, G
Sch \"a fer, T. M.; Settanni, G. Data reweighting in metadynamics simulations. J. Chem. Theory Comput. 2020, 16, 2042--2052
2020
-
[64]
Paszke, A. et al. Proceedings of the 33rd International Conference on Neural Information Processing Systems; Curran Associates Inc.: Red Hook, NY, USA, 2019
2019
-
[65]
Murphy, K. P. Probabilistic Machine Learning: An introduction; MIT Press, 2022
2022
-
[66]
Kingma, D. P.; Ba, J. Adam: A Method for Stochastic Optimization. 2014; https://arxiv.org/abs/1412.6980 mcitethebibliography arxiv.bib0000664000000000000000000045475315174703133011413 0ustar rootroot@article Farmahini2015, doi = 10.1016/j.carbon.2014.11.013 , url = https://doi.org/10.1016/j.carbon.2014.11.013 , year = 2015 , month = mar, publisher = Elsev...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.