Recognition: unknown
Probability Distribution Analysis of the Cascaded Variational Quantum Eigensolver
Pith reviewed 2026-05-09 19:15 UTC · model grok-4.3
The pith
Analyzing probability distributions during cascaded variational quantum eigensolver runs selects guiding states that deliver accurate molecular ground-state energies with minimal quantum resources.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
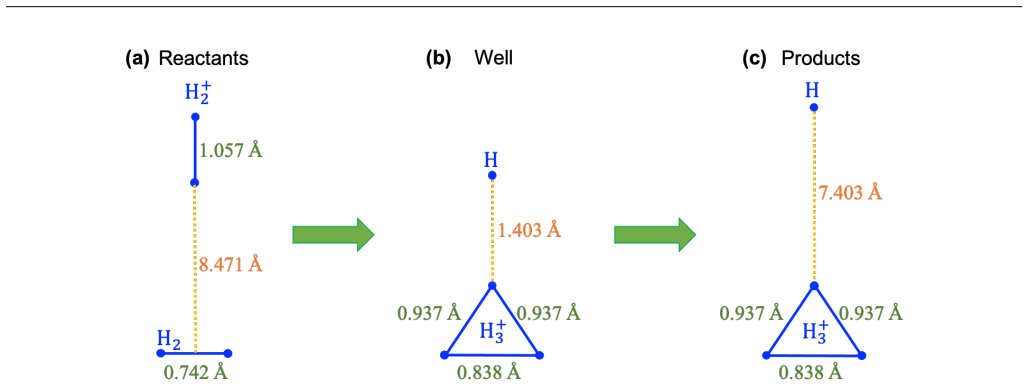
The cascaded variational quantum eigensolver circumvents iterative quantum-classical communication while leaving complete freedom in the choice of guiding state. Not every guiding state yields both accuracy and resource efficiency. A process based on trapezoidal-state preparation, followed by analysis of state probability distributions at different CVQE stages, identifies the optimal guiding-state parameters for given resource constraints. The process is shown to produce accurate electronic energies along the minimal-energy path of the bimolecular reaction H₂ + H₂⁺ → H₃⁺ + H on NISQ hardware.
What carries the argument
Trapezoidal-state preparation followed by probability-distribution analysis at successive CVQE stages, used to select guiding-state parameters.
If this is right
- Accurate ground-state energies for many-electron systems become obtainable on NISQ hardware without repeated quantum-classical iterations.
- Resource consumption is minimized by rejecting guiding states whose probability distributions indicate poor overlap or high variance at early stages.
- Guiding-state parameters can be tuned directly from distribution features rather than exhaustive search for each new molecular problem.
- Electronic energies along reaction paths can be computed reliably once the distribution-based selection rule is fixed for a given qubit and gate budget.
Where Pith is reading between the lines
- If the distribution patterns prove similar across related reactions, the same selection rule could be reused without re-optimization.
- The method may reduce the number of quantum circuit executions needed to reach chemical accuracy by discarding unsuitable guiding states before full variational optimization.
- Connections to other state-preparation circuits in variational algorithms could allow the trapezoidal approach to be adapted for different ansatze or hardware noise profiles.
Load-bearing premise
The observed probability distributions from trapezoidal preparation reliably flag guiding-state parameters that remain accurate for any chosen resource budget and for reactions beyond the single bimolecular case examined.
What would settle it
Apply the selected guiding-state parameters to a second, chemically distinct reaction under the same resource limits and compare the resulting energies against classical benchmarks; systematic deviation would falsify the claim that the distributions identify generally reliable parameters.
Figures







read the original abstract
The cascaded variational quantum eigensolver (CVQE) circumvents the need for iterative communication between the quantum and classical processing units that is necessary in the conventional VQE algorithm. While CVQE offers complete freedom to choose the guiding state as input, not all guiding states suffice for solution accuracy, as well as resource efficiency. Our work presents a process based on trapezoidal-state preparation for selecting guiding states that yield accurate many-electron ground-state solutions with minimal resource consumption. By analyzing the state probability distributions at different stages of the CVQE calculations, we determine the optimal guiding-state parameters for given resource constraints. We demonstrate the process by comparing electronic energies along the minimal-energy path for a prototypical bimolecular reaction, $\mathrm{H}_2 + \mathrm{H}_2^+ \rightarrow \mathrm{H}_3^+ + \mathrm{H}$, using Noisy Intermediate-Scale Quantum (NISQ) computing.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a trapezoidal-state preparation procedure within the cascaded variational quantum eigensolver (CVQE) framework. By examining the probability distributions of the prepared states at successive CVQE stages, the authors claim to identify guiding-state parameters that simultaneously achieve chemical accuracy for many-electron ground states and minimize quantum resource consumption. The method is illustrated on the single bimolecular reaction H₂ + H₂⁺ → H₃⁺ + H along its minimal-energy path using NISQ hardware.
Significance. If the probability-distribution criterion proves robust, the approach could reduce the trial-and-error overhead of guiding-state selection in CVQE, which is relevant for near-term quantum chemistry simulations on NISQ devices. The paper does not, however, supply machine-checked proofs, reproducible code repositories, or parameter-free derivations that would strengthen its long-term impact.
major comments (2)
- [Demonstration / Results section (implicit in abstract and § on numerical experiments)] The central claim that the probability-distribution analysis 'reliably identifies guiding-state parameters that produce accurate solutions under arbitrary resource constraints' (abstract) is supported only by results for the single reaction H₂ + H₂⁺ → H₃⁺ + H. No additional molecules, active-space sizes, or Hamiltonian variations are presented to test whether the same distribution-based selection rule recovers chemical accuracy when the resource budget or the electronic structure changes. This limits the procedure from an empirical observation to a validated general method.
- [Abstract and numerical results] The abstract states that the method yields 'accurate many-electron ground-state solutions with minimal resource consumption,' yet the provided summary supplies no numerical energies, error bars, wall-clock times, or gate-count comparisons against standard VQE or other CVQE variants. Without these quantitative baselines (e.g., in a results table), it is impossible to verify that the selected parameters indeed satisfy the claimed accuracy-resource trade-off.
minor comments (2)
- [Methods] Notation for the trapezoidal-state preparation and the probability-distribution metric is introduced without an explicit equation or pseudocode block, making it difficult to reproduce the selection procedure from the text alone.
- [Analysis procedure] The manuscript would benefit from a clear statement of the precise threshold or figure of merit (e.g., overlap or energy variance) used to declare a guiding state 'optimal' from the probability distribution.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We respond point-by-point to the major comments below. Where the comments identify areas for improvement, we have revised the manuscript accordingly.
read point-by-point responses
-
Referee: [Demonstration / Results section (implicit in abstract and § on numerical experiments)] The central claim that the probability-distribution analysis 'reliably identifies guiding-state parameters that produce accurate solutions under arbitrary resource constraints' (abstract) is supported only by results for the single reaction H₂ + H₂⁺ → H₃⁺ + H. No additional molecules, active-space sizes, or Hamiltonian variations are presented to test whether the same distribution-based selection rule recovers chemical accuracy when the resource budget or the electronic structure changes. This limits the procedure from an empirical observation to a validated general method.
Authors: We agree that the numerical demonstration is performed on a single prototypical reaction. The reaction was selected because it features a change in electron number and active-space dimension along the minimal-energy path, providing a non-trivial test of guiding-state selection. The trapezoidal preparation and probability-distribution criterion are formulated without reference to any specific molecular Hamiltonian and are therefore intended to be general. In the revised manuscript we have added an explicit discussion of the method's scope and limitations, clarifying that the present work constitutes a proof-of-principle demonstration rather than an exhaustive validation across chemical space. revision: partial
-
Referee: [Abstract and numerical results] The abstract states that the method yields 'accurate many-electron ground-state solutions with minimal resource consumption,' yet the provided summary supplies no numerical energies, error bars, wall-clock times, or gate-count comparisons against standard VQE or other CVQE variants. Without these quantitative baselines (e.g., in a results table), it is impossible to verify that the selected parameters indeed satisfy the claimed accuracy-resource trade-off.
Authors: The full manuscript already contains the requested quantitative information in the Numerical Experiments section, including tables of electronic energies, deviations from chemical accuracy, hardware error bars, and explicit gate-count and circuit-depth comparisons with standard VQE. To make these baselines immediately visible, we have revised the abstract to state the achieved accuracy (chemical accuracy within 1.6 mHa) and have inserted a consolidated summary table at the beginning of the results section. revision: yes
Circularity Check
No circularity; empirical distribution analysis selects parameters without reducing to input by construction
full rationale
The paper presents an empirical procedure: trapezoidal-state preparation followed by analysis of probability distributions at CVQE stages to choose guiding-state parameters that minimize resource use while achieving accuracy. This is demonstrated on the H2 + H2+ → H3+ + H reaction. No equations, fitted parameters, or self-citations are invoked that make the selection process equivalent to its own inputs by definition. The central claim remains an observational mapping from observed distributions to parameter choices, not a tautological renaming or self-referential fit. Limitation to a single reaction affects generalizability but does not create circularity in the derivation chain.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Adiabatic Guiding State The parametersℏΩ = 10Ha andK= 500were chosen such that both discretized adiabatic conditions are satis- fied on both ends by a healthy margin, while maximizing the number of steps. This combination yielded an en- ergy errorE error of0.001eV for the produced trapezoidal state and0.009eV for the guiding state relative toE g, both wel...
-
[2]
Ideal Trapezoidal State The two discretized adiabatic conditions in Eq.(10) are not necessarily of equal importance. The purpose of the right condition is to prevent excitations during the adiabatic process; a sufficient choice in our calcu- lations for the trapezoidal-state preparation isℏΩ = 1.0Ha. The advantage of relaxing this condition slightly is th...
-
[3]
This means that the practical bottleneck in our approach is the circuit depth
Single-step Guiding State A challenge as we perform calculations on larger sys- tems is that the number of interactions also increases, generally with the fourth power of the system size. This means that the practical bottleneck in our approach is the circuit depth. One question is then how much we can control the circuit depth using the number of stepsK....
-
[4]
To see how real- world system noise impacts our calculations, we have also performed calculations on theibm_aachenquantum computer
Quantum Processing Up to this point, the presented results have been ob- tained using a noiseless IBM simulator. To see how real- world system noise impacts our calculations, we have also performed calculations on theibm_aachenquantum computer. The circuit depth has again been kept mini- mal by choosing a single-step guiding state,K= 1, drop- ping system ...
-
[5]
A. Y. Kitaev, Quantum measurements and the abelian stabilizer problem, arXiv preprint quant-ph/9511026 (1995)
work page internal anchor Pith review arXiv 1995
-
[6]
Van Dam, M
W. Van Dam, M. Mosca, and U. Vazirani, How power- ful is adiabatic quantum computation?, inProceedings 42nd IEEE symposium on foundations of computer sci- ence(IEEE, 2001) pp. 279–287
2001
-
[7]
Preskill, Quantum computing in the nisq era and be- yond, Quantum2, 79 (2018)
J. Preskill, Quantum computing in the nisq era and be- yond, Quantum2, 79 (2018)
2018
-
[8]
Peruzzo, J
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Distribution Statement A. Approved for public release: distribution is unlimited. 7 FIG. 10. Sampling counts from the QPU,ibm_aachen, across all the 256 Fock states|n⟩for the well geometry, with|S|= 106. The red bars represent the Fock states that appear in the ground state, while the blue bars repres...
2014
-
[9]
J. R. McClean, J. Romero, R. Babbush, and A. Aspuru- Guzik, The theory of variational hybrid quantum- classical algorithms, New Journal of Physics18, 023023 (2016)
2016
-
[10]
P. J. O’Malley, R. Babbush, I. D. Kivlichan, J. Romero, J. R. McClean, R. Barends, J. Kelly, P. Roushan, A. Tranter, N. Ding,et al., Scalable quantum simula- tion of molecular energies, Physical Review X6, 031007 (2016)
2016
-
[11]
Kandala, A
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, Hardware- efficient variational quantum eigensolver for small moleculesandquantummagnets,nature549,242(2017)
2017
-
[12]
D. Wang, O. Higgott, and S. Brierley, Accelerated varia- tional quantum eigensolver, Physical review letters122, 140504 (2019)
2019
-
[13]
G. A. Quantum, Collaborators*†, F. Arute, K. Arya, R. Babbush, D. Bacon, J. C. Bardin, R. Barends, S. Boixo, M. Broughton, B. B. Buckley,et al., Hartree- fock on a superconducting qubit quantum computer, Sci- ence369, 1084 (2020)
2020
-
[14]
J. F. Gonthier, M. D. Radin, C. Buda, E. J. Doskocil, C. M. Abuan, and J. Romero, Measurements as a road- blocktonear-termpracticalquantumadvantageinchem- istry: Resource analysis, Physical Review Research4, 033154 (2022)
2022
-
[15]
McArdle, S
S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin, and X. Yuan, Quantum computational chemistry, Re- views of Modern Physics92, 015003 (2020)
2020
-
[16]
Cerezo, A
M. Cerezo, A. Arrasmith, R. Babbush, S. C. Benjamin, S. Endo, K. Fujii, J. R. McClean, K. Mitarai, X. Yuan, L. Cincio,et al., Variational quantum algorithms, Nature Reviews Physics3, 625 (2021)
2021
-
[17]
Head-Marsden, J
K. Head-Marsden, J. Flick, C. J. Ciccarino, and P. Narang, Quantum information and algorithms for cor- related quantum matter, Chemical Reviews121, 3061 (2020). Distribution Statement A. Approved for public release: distribution is unlimited. 8
2020
-
[18]
Gunlycke, C
D. Gunlycke, C. S. Hellberg, and J. P. Stenger, Cascaded variational quantum eigensolver algorithm, Physical Re- view Research6, 013238 (2024)
2024
-
[19]
Guided sampling ansätzes for variational quantum computing,
D. Gunlycke, J. Stenger, A. Maksymov, A. Kaushik, M. Roetteler, and C. S. Hellberg, Guided sampling ansatzes for variational quantum computing, arXiv preprint arXiv:2508.13926 (2025)
-
[20]
Hybrid vqe-cvqe algorithm using diabatic state preparation,
J. Stenger, C. S. Hellberg, and D. Gunlycke, Hybrid vqe- cvqe algorithm using diabatic state preparation, arXiv preprint arXiv:2512.04801 (2025)
-
[21]
Herbst and W
E. Herbst and W. Klemperer, The formation and deple- tion of molecules in dense interstellar clouds, Astrophysi- cal Journal, Vol. 185, pp. 505-534 (1973)185, 505 (1973)
1973
-
[22]
W. D. Watson, The rate of formation of interstellar molecules by ion-molecule reactions, Astrophysical Jour- nal, vol. 183, p. L17183, L17 (1973)
1973
-
[23]
Prasad and A
S. Prasad and A. Tan, The jovian ionosphere, Geophysi- cal Research Letters1, 337 (1974)
1974
-
[24]
Oka, Interstellar h3+, Proceedings of the National Academy of Sciences103, 12235 (2006)
T. Oka, Interstellar h3+, Proceedings of the National Academy of Sciences103, 12235 (2006)
2006
-
[25]
Klemperer, Interstellar chemistry, Proceedings of the National Academy of Sciences103, 12232 (2006)
W. Klemperer, Interstellar chemistry, Proceedings of the National Academy of Sciences103, 12232 (2006)
2006
-
[26]
Oka, Interstellar h3+, Chemical Reviews113, 8738 (2013)
T. Oka, Interstellar h3+, Chemical Reviews113, 8738 (2013)
2013
-
[27]
Farhi, J
E. Farhi, J. Goldstone, S. Gutmann, J. Lapan, A. Lund- gren, and D. Preda, A quantum adiabatic evolution al- gorithm applied to random instances of an np-complete problem, Science292, 472 (2001)
2001
-
[28]
V. Smelyanskiy, U. Toussaint, and D. Timucin, Simu- lations of the adiabatic quantum optimization for the set partition problem, arXiv preprint quant-ph/0112143 (2001)
-
[29]
M. Suzuki, Generalized trotter’s formula and systematic approximants of exponential operators and inner deriva- tions with applications to many-body problems, Commu- nications in Mathematical Physics51, 183 (1976)
1976
-
[30]
Suzuki, On the convergence of exponential operators—the zassenhaus formula, bch formula and systematic approximants, Communications in Mathematical Physics57, 193 (1977)
M. Suzuki, On the convergence of exponential operators—the zassenhaus formula, bch formula and systematic approximants, Communications in Mathematical Physics57, 193 (1977)
1977
-
[31]
M. Suzuki, Decomposition formulas of exponential op- erators and lie exponentials with some applications to quantum mechanics and statistical physics, Journal of mathematical physics26, 601 (1985)
1985
-
[32]
Huyghebaert and H
J. Huyghebaert and H. De Raedt, Product formula meth- ods for time-dependent schrodinger problems, Journal of Physics A: Mathematical and General23, 5777 (1990)
1990
-
[33]
G.Mills, H.Jónsson,andG.K.Schenter,Reversiblework transition state theory: application to dissociative ad- sorption of hydrogen, Surface Science324, 305 (1995)
1995
-
[34]
Jónsson, G
H. Jónsson, G. Mills, andK. W.Jacobsen, Nudged elastic band method for finding minimum energy paths of tran- sitions, inClassical and quantum dynamics in condensed phase simulations(World Scientific, 1998) pp. 385–404
1998
-
[35]
W. J. Hehre, R. F. Stewart, and J. A. Pople, Self- consistent molecular-orbital methods. i. use of gaussian expansions of slater-type atomic orbitals, The Journal of Chemical Physics51, 2657 (1969)
1969
-
[36]
D. G. Smith, L. A. Burns, A. C. Simmonett, R. M. Par- rish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Di Remigio, A. Alenaizan,et al., Psi4 1.4: Open- source software for high-throughput quantum chemistry, The Journal of chemical physics152(2020)
2020
-
[37]
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus,et al., The atomic sim- ulation environment—a python library for working with atoms, Journal of Physics: Condensed Matter29, 273002 (2017)
2017
-
[38]
Jordan and E
P. Jordan and E. Wigner, Über das paulische äquivalen- zverbot, Zeitschrift für Physik47, 631 (1928)
1928
-
[39]
Krenos, K
J. Krenos, K. Lehmann, J. Tully, P. Hierl, and G. Smith, Crossed-beam study of the reactions of h+ 2 with d2 and d+ 2 with h2, Chemical Physics16, 109 (1976)
1976
-
[40]
Stine and J
J. Stine and J. Muckerman, Charge exchange and chemi- cal reaction in the h2++ h2 system. i. characterization of the potential energy surfaces and nonadiabatic regions, The Journal of Chemical Physics68, 185 (1978)
1978
-
[41]
Pollard, L
J. Pollard, L. Johnson, D. Lichtin, and R. Cohen, State- selected reactive scattering. i. h+ 2+ h2→h+ 3+ h, The Journal of chemical physics95, 4877 (1991)
1991
-
[42]
A. Javadi-Abhari, M. Treinish, K. Krsulich, C. J. Wood, J. Lishman, J. Gacon, S. Martiel, P. D. Na- tion, L. S. Bishop, A. W. Cross, B. R. Johnson, and J.M.Gambetta,QuantumcomputingwithQiskit(2024), arXiv:2405.08810 [quant-ph]
work page internal anchor Pith review arXiv 2024
-
[43]
A. M. Childs, Y. Su, M. C. Tran, N. Wiebe, and S. Zhu, Theory of trotter error with commutator scaling, Physi- cal Review X11, 011020 (2021)
2021
-
[44]
Layden, First-order trotter error from a second-order perspective, Physical Review Letters128, 210501 (2022)
D. Layden, First-order trotter error from a second-order perspective, Physical Review Letters128, 210501 (2022)
2022
-
[45]
Suzuki, General theory of fractal path integrals with applications to many-body theories and statistical physics, Journal of mathematical physics32, 400 (1991)
M. Suzuki, General theory of fractal path integrals with applications to many-body theories and statistical physics, Journal of mathematical physics32, 400 (1991)
1991
-
[46]
L. A. Dias and R. B. Faria, Practical decomposition of irreducible representations: Applications to molecular vi- brations and molecular orbitals, Journal of Chemical Ed- ucation97, 2332 (2020). Distribution Statement A. Approved for public release: distribution is unlimited. 9 Appendix I. ADIABA TIC CONDITION FOR DISCRETIZED ADIABA TIC EVOLUTION Here we ...
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.