Recognition: unknown
Stochastic Cluster Expansion for Excited State Energies
Pith reviewed 2026-05-09 17:50 UTC · model grok-4.3
The pith
Excitation energies are reconstructed as a hierarchy of orbital cluster contributions using an exactly treated minimal frontier subspace plus stochastic sampling of the rest.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By expressing excitation energies as a hierarchy of orbital-space cluster contributions, the method reconstructs singlet-triplet gaps from reduced-rank calculations: a minimal frontier chemical subspace is treated exactly while the remaining orbital environment is sampled stochastically. On charge-transfer complexes and polyacenes the resulting gaps agree with full-system references and converge with low-order terms, eliminating the need for large or preselected active spaces.
What carries the argument
The hierarchy of orbital-space cluster contributions to the excitation energy difference, obtained by exact treatment of a minimal frontier chemical subspace combined with stochastic sampling of the remaining orbitals.
If this is right
- Singlet-triplet gaps on charge-transfer complexes and polyacenes match full-system reference values.
- The expansion converges already at low cluster orders.
- The framework is systematically improvable by adding higher-order cluster terms.
- No large or chemically preselected active space is required.
Where Pith is reading between the lines
- The same cluster hierarchy could be applied to other excitation types such as double excitations or charge-transfer states beyond singlet-triplet gaps.
- For very large molecules the stochastic sampling cost may remain favorable compared with deterministic active-space growth.
- The choice of which orbitals belong to the frontier subspace could be made automatic by an orbital-selection criterion based on occupation or energy.
Load-bearing premise
The excitation energy can be recovered accurately from a hierarchy of calculations on a small exactly solved orbital subspace plus stochastic estimates of the environment without missing essential correlations.
What would settle it
A direct comparison on a molecule where both the new method and a full-system exact diagonalization can be performed, showing a singlet-triplet gap difference larger than the agreement reported for the tested systems.
Figures


read the original abstract
Excited-state electronic structure in strongly correlated systems remains challenging due to the exponential scaling of the many-body Hilbert space and the difficulty of constructing systematically controlled active spaces. Building on the stochastic cluster expansion (SCE) framework previously developed for ground-state correlation energies, we extend the formalism to excitation gaps by expressing energy differences directly as a hierarchy of orbital-space cluster contributions. In this formulation, excitation energies are reconstructed from reduced-rank calculations involving a minimal frontier chemical subspace (FCS), treated exactly, together with stochastic sampling of the remaining orbital environment. This approach eliminates the need for large or chemically preselected active spaces. We demonstrate the method on charge-transfer complexes and polyacenes, where accurate singlet-triplet gaps are obtained that agree with full-system results. The method converges with low-order cluster terms and provides a systematically improvable framework for excited states in correlated systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript extends the stochastic cluster expansion (SCE) framework from ground states to excitation energies by expressing singlet-triplet gaps directly as a hierarchy of orbital cluster contributions. A minimal frontier chemical subspace is treated exactly while the remaining orbital environment is sampled stochastically, eliminating the need for large active spaces. Benchmarks on charge-transfer complexes and polyacenes show that the method converges with low-order terms and reproduces full-system reference values to within chemical accuracy.
Significance. If the central claims hold, the work supplies a systematically improvable, parameter-free route to excited-state energies in correlated systems that avoids the exponential cost of full configuration interaction or the arbitrariness of active-space selection. The reported convergence behavior and agreement with full-system results on chemically relevant benchmarks constitute a concrete strength that could be extended to other excitation types.
minor comments (2)
- [Abstract] The abstract states agreement with full-system results but does not report quantitative error metrics or statistical uncertainties arising from the stochastic sampling; adding these (e.g., mean absolute deviations and standard errors) would make the claim more precise.
- [Results and Discussion] In the benchmark sections, the convergence plots and tables would benefit from explicit tabulation of the stochastic sampling parameters (number of samples, variance estimates) alongside the reported energies to allow readers to assess the statistical reliability of the low-order truncation.
Simulated Author's Rebuttal
We thank the referee for the positive summary, significance assessment, and recommendation for minor revision. The referee accurately captures the extension of stochastic cluster expansion to excitation energies via a minimal frontier chemical subspace treated exactly and stochastic sampling of the orbital environment, along with the benchmarks on charge-transfer complexes and polyacenes. No specific major comments were provided in the report.
Circularity Check
Minor self-citation to prior ground-state SCE; excited-state derivation is independent
full rationale
The paper builds on a previously developed SCE framework for ground-state energies but formulates the extension to excitation gaps by directly expressing energy differences as a hierarchy of orbital-space cluster contributions, with a minimal frontier chemical subspace treated exactly plus stochastic sampling of the environment. This difference-based reconstruction is presented as a new, self-contained formulation without reducing to fitted parameters, self-referential definitions, or load-bearing self-citations that would force the result by construction. Benchmarks on charge-transfer complexes and polyacenes demonstrate convergence to full-system references with low-order terms, confirming the chain does not collapse to its inputs. The single self-citation to the ground-state method is not load-bearing for the central excited-state claims.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Excitation energies can be expressed as a hierarchy of orbital-space cluster contributions from a minimal exact subspace plus stochastic environment.
Reference graph
Works this paper leans on
-
[1]
Kasha, Characterization of electronic transitions in complex molecules, Discussions of the Faraday society9, 14 (1950)
M. Kasha, Characterization of electronic transitions in complex molecules, Discussions of the Faraday society9, 14 (1950)
1950
-
[2]
Kasha, Paths of molecular excitation, Radiation re- search supplement2, 243 (1960)
M. Kasha, Paths of molecular excitation, Radiation re- search supplement2, 243 (1960)
1960
-
[3]
J. R. Lakowicz,Principles of Fluorescence Spectroscopy, 3rd ed. (Springer, Singapore, 2006)
2006
-
[4]
Y. Ma, M. Rohlfing, and C. Molteni, Modeling the ex- cited states of biological chromophores within many- body green’s function theory, Journal of Chemical The- ory and Computation6, 257 (2010), pMID: 26614336, https://doi.org/10.1021/ct900528h
-
[5]
P. M. Zimmerman, Z. Zhang, and C. B. Musgrave, Sin- glet fission in pentacene through multi-exciton quantum states, Nature Chemistry2, 648 (2010)
2010
-
[6]
Uoyama, K
H. Uoyama, K. Goushi, K. Shizu, H. Nomura, and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature492, 234 (2012)
2012
-
[7]
David Sherrill and H
C. David Sherrill and H. F. Schaefer, The configuration interaction method: Advances in highly correlated ap- proaches, inAdvances in Quantum Chemistry(Elsevier,
-
[8]
J. J. Eriksen, The shape of full configuration interaction to come, The Journal of Physical Chemistry Letters12, 418 (2020)
2020
-
[9]
C. U. Ibeji and D. Ghosh, Singlet–triplet gaps in poly- acenes: a delicate balance between dynamic and static correlations investigated by spin–flip methods, Phys. Chem. Chem. Phys.17, 9849 (2015)
2015
-
[10]
Hachmann, J
J. Hachmann, J. J. Dorando, M. Avil´ es, and G. K.-L. Chan, The radical character of the acenes: A density ma- trix renormalization group study, The Journal of Chem- ical Physics127, 134309 (2007)
2007
-
[11]
J. Shee, E. J. Arthur, S. Zhang, D. R. Reichman, and R. A. Friesner, Singlet-triplet energy gaps of organic biradicals and polyacenes with auxiliary-field quantum monte carlo, Journal of Chemical Theory and Computa- tion15, 4924 (2019), pMID: 31381324
2019
-
[12]
Sharma, V
P. Sharma, V. Bernales, S. Knecht, D. G. Truhlar, and L. Gagliardi, Density matrix renormalization group pair- 7 density functional theory (dmrg-pdft): singlet–triplet gaps in polyacenes and polyacetylenes, Chem. Sci.10, 1716 (2019)
2019
-
[13]
Ghosh, C
S. Ghosh, C. J. Cramer, D. G. Truhlar, and L. Gagliardi, Generalized-active-space pair-density functional theory: an efficient method to study large, strongly correlated, conjugated systems, Chemical science8, 2741 (2017)
2017
- [14]
-
[15]
E. Kolodzeiski and C. J. Stein, Automated, consis- tent, and even-handed selection of active orbital spaces for quantum embedding, Journal of Chemical Theory and Computation19, 6643 (2023), pMID: 37775093, https://doi.org/10.1021/acs.jctc.3c00653
-
[16]
He and F
N. He and F. A. Evangelista, A zeroth-order active-space frozen-orbital embedding scheme for multireference cal- culations, The Journal of Chemical Physics152, 094107 (2020)
2020
-
[17]
Muechler, D
L. Muechler, D. I. Badrtdinov, A. Hampel, J. Cano, M. R¨ osner, and C. E. Dreyer, Quantum embedding meth- ods for correlated excited states of point defects: Case studies and challenges, Phys. Rev. B105, 235104 (2022)
2022
-
[18]
G. Weng, M. Romanova, A. Apelian, H. Song, and V. Vlˇ cek, Reduced scaling of optimal regional or- bital localization via sequential exhaustion of the single-particle space, Journal of Chemical Theory and Computation18, 4960 (2022), pMID: 35817013, https://doi.org/10.1021/acs.jctc.2c00315
-
[19]
G. Weng and V. Vlˇ cek, Efficient treatment of molecular excitations in the liquid phase environment via stochastic many-body theory, The Journal of Chemical Physics155, 054104 (2021), https://pubs.aip.org/aip/jcp/article- pdf/doi/10.1063/5.0058410/14111299/054104 1 online.pdf
-
[20]
R. J. Bartlett and M. Musia l, Coupled-cluster theory in quantum chemistry, Reviews of Modern Physics79, 291–352 (2007)
2007
-
[21]
Baiardi and M
A. Baiardi and M. Reiher, The density matrix renormal- ization group in chemistry and molecular physics: Recent developments and new challenges, J. Chem. Phys.152, 040903 (2020)
2020
-
[22]
S. R. White, Density matrix formulation for quantum renormalization groups, Phys. Rev. Lett.69, 2863 (1992)
1992
-
[23]
R. Olivares-Amaya, W. Hu, N. Nakatani, S. Sharma, J. Yang, and G. K.-L. Chan, The ab-initio density ma- trix renormalization group in practice, The Journal of Chemical Physics142, 10.1063/1.4905329 (2015)
-
[24]
E. R. Sayfutyarova, Q. Sun, G. K.-L. Chan, and G. Knizia, Automated construction of molecular active spaces from atomic valence orbitals, Journal of chemical theory and computation13, 4063 (2017)
2017
-
[25]
J. J. Bao and D. G. Truhlar, Automatic active space se- lection for calculating electronic excitation energies based on high-spin unrestricted hartree–fock orbitals, Journal of Chemical Theory and Computation15, 5308 (2019)
2019
-
[26]
Sakuma, P
R. Sakuma, P. Werner, and F. Aryasetiawan, Electronic structure of srvo 3 withingw+dmft, Phys. Rev. B88, 235110 (2013)
2013
-
[27]
Aryasetiawan, J
F. Aryasetiawan, J. M. Tomczak, T. Miyake, and R. Sakuma, Downfolded self-energy of many-electron sys- tems, Phys. Rev. Lett.102, 176402 (2009)
2009
-
[28]
Miyake, F
T. Miyake, F. Aryasetiawan, and M. Imada, Ab initio procedure for constructing effective models of correlated materials with entangled band structure, Phys. Rev. B 80, 155134 (2009)
2009
-
[29]
Romanova, G
M. Romanova, G. Weng, A. Apelian, and V. Vlˇ cek, Dy- namical downfolding for localized quantum states, npj Computational Materials9, 126 (2023)
2023
-
[30]
Szalay, M
S. Szalay, M. Pfeffer, V. Murg, G. Barcza, F. Verstraete, R. Schneider, and ¨Ors Legeza, Tensor product meth- ods and entanglement optimization forab initioquantum chemistry, Int. J. Quant. Chem.115, 1342 (2015)
2015
-
[31]
Legeza, J
¨O. Legeza, J. R¨ oder, and B. Hess, Phys. Rev. B67, 125114 (2003)
2003
-
[32]
O. Legeza and J. S´ olyom, Optimizing the density- matrix renormalization group method using quantum information entropy, Phys. Rev. B68, 10.1103/phys- revb.68.195116 (2003)
-
[33]
J. Brabec, J. Brandejs, K. Kowalski, S. Xantheas, O. Legeza, and L. Veis, Massively parallel quantum chemical density matrix renormalization group method, Journal of Computational Chemistry42, 534 (2021), https://onlinelibrary.wiley.com/doi/pdf/10.1002/jcc.26476
-
[34]
G. K.-L. Chan and S. Sharma, The density matrix renor- malization group in quantum chemistry, Annu. Rev. Phys. Chem.62, 465 (2011)
2011
-
[35]
B. Eskridge, H. Krakauer, and S. Zhang, Local em- bedding and effective downfolding in the auxiliary-field quantum monte carlo method, Journal of Chemical The- ory and Computation15, 3949 (2019), pMID: 31244125, https://doi.org/10.1021/acs.jctc.8b01244
- [36]
-
[37]
Malmqvist and B
P.-˚A. Malmqvist and B. O. Roos, The casscf state inter- action method, Chemical physics letters155, 189 (1989)
1989
-
[38]
Helmich-Paris, Benchmarks for electronically excited states with casscf methods, Journal of Chemical Theory and Computation15, 4170 (2019)
B. Helmich-Paris, Benchmarks for electronically excited states with casscf methods, Journal of Chemical Theory and Computation15, 4170 (2019)
2019
-
[39]
Olsen, The casscf method: A perspective and commen- tary, International Journal of Quantum Chemistry111, 3267 (2011)
J. Olsen, The casscf method: A perspective and commen- tary, International Journal of Quantum Chemistry111, 3267 (2011)
2011
-
[40]
Y. Yang, E. R. Davidson, and W. Yang, Nature of ground and electronic excited states of higher acenes, Proceed- ings of the National Academy of Sciences113, E5098 (2016)
2016
-
[41]
Z. Qu, D. Zhang, C. Liu, and Y. Jiang, Open-shell ground state of polyacenes: a valence bond study, The Journal of Physical Chemistry A113, 7909 (2009)
2009
-
[42]
D. Drwal, P. Beran, M. Hapka, M. Modrzejewski, A. Sok´ o l, L. Veis, and K. Pernal, Efficient adiabatic con- nection approach for strongly correlated systems: Appli- cation to singlet–triplet gaps of biradicals, The Journal of Physical Chemistry Letters13, 4570 (2022), pMID: 35580342, https://doi.org/10.1021/acs.jpclett.2c00993
-
[43]
Li Manni, R
G. Li Manni, R. K. Carlson, S. Luo, D. Ma, J. Olsen, D. G. Truhlar, and L. Gagliardi, Multiconfiguration pair- density functional theory, Journal of chemical theory and computation10, 3669 (2014)
2014
-
[44]
Pernal, Electron correlation from the adiabatic con- nection for multireference wave functions, Physical re- view letters120, 013001 (2018)
K. Pernal, Electron correlation from the adiabatic con- nection for multireference wave functions, Physical re- view letters120, 013001 (2018). 8
2018
-
[45]
Beran, M
P. Beran, M. Matousek, M. Hapka, K. Pernal, and L. Veis, Density matrix renormalization group with dy- namical correlation via adiabatic connection, Journal of Chemical Theory and Computation17, 7575 (2021)
2021
-
[46]
Andersson, P.-˚A
K. Andersson, P.-˚A. Malmqvist, and B. O. Roos, Second- order perturbation theory with a complete active space self-consistent field reference function, The Journal of chemical physics96, 1218 (1992)
1992
-
[47]
Angeli, R
C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger, and J.-P. Malrieu, Introduction of n-electron valence states for multireference perturbation theory, The Jour- nal of Chemical Physics114, 10252 (2001)
2001
-
[48]
H. Ma, N. Sheng, M. Govoni, and G. Galli, Quantum embedding theory for strongly correlated states in mate- rials, J. Chem. Theory Comput.17, 2116 (2021)
2021
-
[49]
Dvorak and P
M. Dvorak and P. Rinke, Dynamical configuration inter- action: Quantum embedding that combines wave func- tions and green’s functions, Phys. Rev. B99, 115134 (2019)
2019
-
[50]
Dvorak, D
M. Dvorak, D. Golze, and P. Rinke, Quantum embedding theory in the screened coulomb interaction: Combin- ing configuration interaction withgw/BSE, Phys. Rev. Mater.3, 070801 (2019)
2019
-
[51]
Sheng, C
N. Sheng, C. Vorwerk, M. Govoni, and G. Galli, Green’s function formulation of quantum defect embedding the- ory, J. Chem. Theory Comput.18, 3512 (2022)
2022
-
[52]
Chang, E
Y. Chang, E. G. van Loon, B. Eskridge, B. Busemeyer, M. A. Morales, C. E. Dreyer, A. J. Millis, S. Zhang, T. O. Wehling, L. K. Wagner,et al., Downfolding from ab initio to interacting model hamiltonians: comprehensive anal- ysis and benchmarking of the dft+ crpa approach, npj Computational Materials10, 129 (2024)
2024
-
[53]
Chang, S
Y. Chang, S. Joshi, and L. K. Wagner, Renormalized den- sity matrix downfolding: A rigorous framework in learn- ing emergent models from ab initio many-body calcula- tions, Physical Review B110, 195103 (2024)
2024
-
[54]
From Real Materials to Model Hamiltonians With Density Matrix Downfolding,
H. Zheng, H. J. Changlani, K. T. Williams, B. Buse- meyer, and L. K. Wagner, From real materials to model hamiltonians with density matrix downfolding, Frontiers in PhysicsV olume 6 - 2018, 10.3389/fphy.2018.00043 (2018)
-
[55]
P. H. Dederichs, S. Bl¨ ugel, R. Zeller, and H. Akai, Ground states of constrained systems: Application to cerium im- purities, Phys. Rev. Lett.53, 2512 (1984)
1984
-
[56]
Gunnarsson, O
O. Gunnarsson, O. K. Andersen, O. Jepsen, and J. Zaa- nen, Density-functional calculation of the parameters in the anderson model: Application to mn in cdte, Phys. Rev. B39, 1708 (1989)
1989
-
[57]
A. Canestraight, A. Dominic III, Anthony J.and Montoya-Castillo, L. Veis, and V. Vlcek, A stochastic cluster expansion for electronic correlation in large sys- tems, The Journal of Physical Chemistry Letters0, null (0), https://doi.org/10.1021/acs.jpclett.6c00563
-
[58]
G. R. Medders, A. W. G¨ otz, M. A. Morales, P. Bajaj, and F. Paesani, On the representation of many-body inter- actions in water, The Journal of Chemical Physics143, 104102 (2015), https://pubs.aip.org/aip/jcp/article- pdf/doi/10.1063/1.4930194/15502073/104102 1 online.pdf
-
[59]
G. R. Medders, V. Babin, and F. Paesani, A critical assessment of two-body and three-body in- teractions in water, Journal of Chemical Theory and Computation9, 1103 (2013), pMID: 26588754, https://doi.org/10.1021/ct300913g
-
[60]
R. K. Nesbet, Atomic bethe-goldstone equations. iii. cor- relation energies of ground states of be, b, c, n, o, f, and ne, Phys. Rev.175, 2 (1968)
1968
-
[61]
V. Abraham and N. J. Mayhall, Cluster many-body ex- pansion: A many-body expansion of the electron cor- relation energy about a cluster mean field reference, The Journal of Chemical Physics155, 10.1063/5.0057752 (2021)
-
[62]
3 requires that the gap is evaluated between states with (nearly) the same two eigenvectors for every stochastic sampling
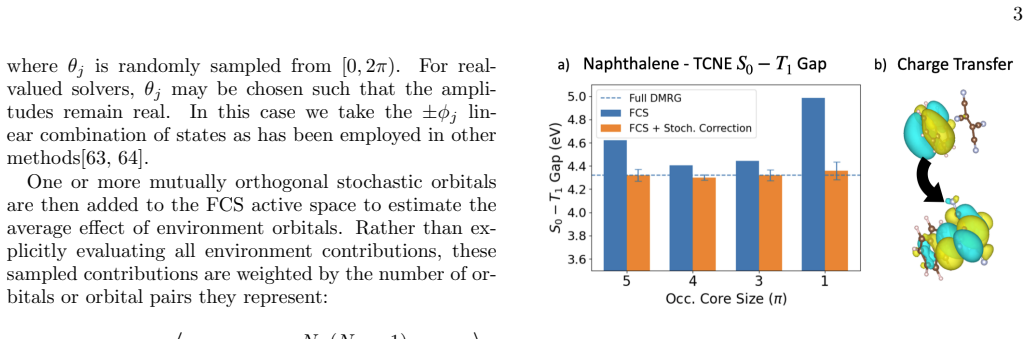
A physically meaningful evaluation Eq. 3 requires that the gap is evaluated between states with (nearly) the same two eigenvectors for every stochastic sampling. When the excitation is not predominantly contained in the FCS, inclusion of stochastic orbitalsϕandϕ ′ may have a large impact on the eigenvectors of the many-body Hamiltonian, rendering the requ...
-
[63]
Canestraight, X
A. Canestraight, X. Lei, K. Z. Ibrahim, and V. Vlˇ cek, Efficient quasiparticle determination beyond the diag- onal approximation via random compression, Journal of Chemical Theory and Computation20, 551 (2024), pMID: 38175913
2024
-
[64]
Apelian, A
A. Apelian, A. Canestraight, S. Liu, and V. Vlcek, Delo- calization of quasiparticle moir´ e states in twisted bilayer hbn, Nano Letters24, 11882 (2024)
2024
-
[65]
Møller and M
C. Møller and M. S. Plesset, Note on an approximation treatment for many-electron systems, Physical Review 46, 618–622 (1934)
1934
-
[66]
D. N. Dhar, The chemistry of tetracyanoethylene, Chem- ical Reviews67, 611 (1967)
1967
-
[67]
T. Stein, L. Kronik, and R. Baer, Reliable prediction of charge transfer excitations in molecular complexes us- ing time-dependent density functional theory, Journal of the American Chemical Society131, 2818 (2009), pMID: 19239266, https://doi.org/10.1021/ja8087482
-
[68]
N. M. Tubman, C. D. Freeman, D. S. Levine, D. Hait, M. Head-Gordon, and K. B. Whaley, Modern approaches to exact diagonalization and selected configuration inter- action with the adaptive sampling ci method, Journal of chemical theory and computation16, 2139 (2020)
2020
-
[69]
Neuscamman, C
E. Neuscamman, C. Umrigar, and G. K.-L. Chan, Op- timizing large parameter sets in variational quantum monte carlo, Physical Review B—Condensed Matter and Materials Physics85, 045103 (2012)
2012
-
[70]
Cleland, G
D. Cleland, G. H. Booth, and A. Alavi, Communica- tions: Survival of the fittest: Accelerating convergence in full configuration-interaction quantum monte carlo, The Journal of chemical physics132(2010)
2010
-
[71]
R. M. Martin, L. Reining, and D. M. Ceperley,Inter- acting Electrons: Theory and Computational Approaches (Cambridge University Press, 2016)
2016
-
[72]
Kuroda, T
H. Kuroda, T. Amano, I. Ikemoto, and H. Akamatu, Charge-transfer interaction in tetracyanoethylene com- plexes of pyrene and naphthalene, Journal of the Ameri- can Chemical Society89, 6056 (1967)
1967
-
[73]
Blase and C
X. Blase and C. Attaccalite, Charge-transfer excitations in molecular donor-acceptor complexes within the many- body bethe-salpeter approach, Applied Physics Letters 99(2011)
2011
-
[74]
Mei and W
Y. Mei and W. Yang, Charge transfer excitation energies from ground state density functional theory calculations, The Journal of Chemical Physics150(2019)
2019
-
[75]
Kronik, T
L. Kronik, T. Stein, S. Refaely-Abramson, and R. Baer, Excitation gaps of finite-sized systems from optimally tuned range-separated hybrid functionals, Journal of 9 Chemical Theory and Computation8, 1515 (2012)
2012
-
[76]
molmps developers, Scalable,https://gitlab.com/ molmps/scalable(2026), gitLab repository, accessed April 30, 2026
2026
-
[77]
Hajgat´ o, D
B. Hajgat´ o, D. Szieberth, P. Geerlings, F. De Proft, and M. Deleuze, A benchmark theoretical study of the elec- tronic ground state and of the singlet-triplet split of ben- zene and linear acenes, The Journal of chemical physics 131(2009)
2009
-
[78]
Jiang and S
D.-e. Jiang and S. Dai, Electronic ground state of higher acenes, The Journal of Physical Chemistry A112, 332 (2008)
2008
-
[79]
S. D. Pineda Flores and E. Neuscamman, Excited state specific multi-slater jastrow wave functions, The Journal of Physical Chemistry A123, 1487 (2019)
2019
-
[80]
Zhao and E
L. Zhao and E. Neuscamman, An efficient variational principle for the direct optimization of excited states, Journal of chemical theory and computation12, 3436 (2016)
2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.