Recognition: 3 theorem links
· Lean TheoremBenchmarking quantum trial wavefunctions for phaseless auxiliary-field quantum Monte Carlo
Pith reviewed 2026-05-08 19:25 UTC · model grok-4.3
The pith
Adaptive quantum ansatze outperform fixed ones in phaseless auxiliary-field quantum Monte Carlo for strongly correlated molecules while using more compact circuits.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
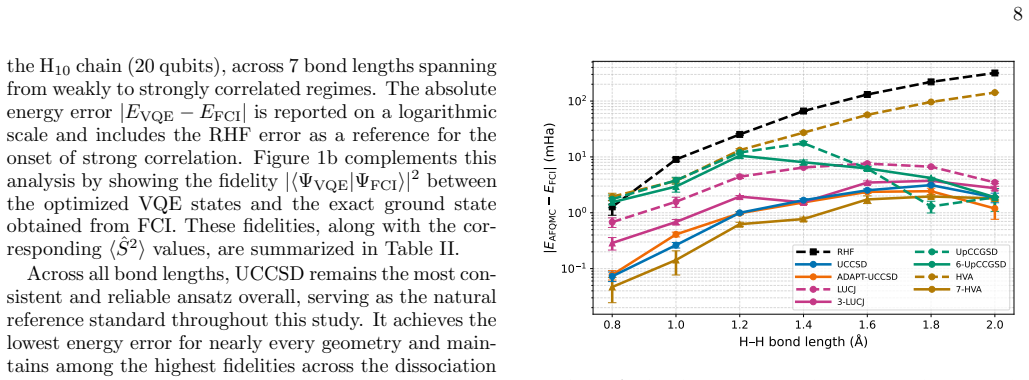
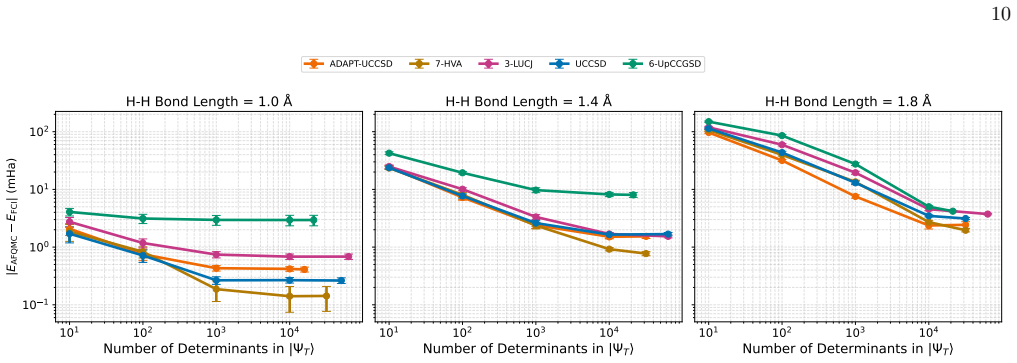
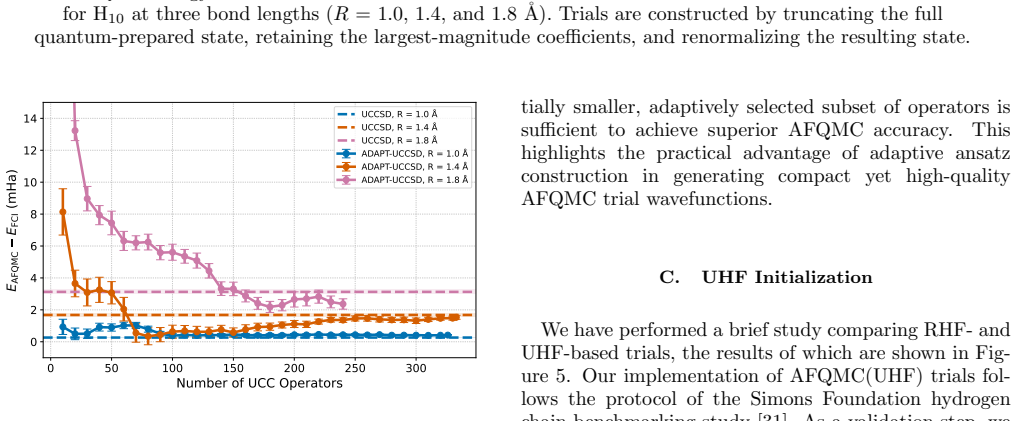
Trial wavefunctions prepared from adaptive ansatze, such as ADAPT-VQE built from the UCCSD operator pool, deliver superior ph-AFQMC projected energies compared with their fixed-ansatz counterparts (UCCSD) in the strongly correlated regime of linear hydrogen chains, while employing substantially more compact quantum circuits. At comparable numbers of variational parameters, different ansatz families produce similar ph-AFQMC accuracies despite large differences in variational energies, optimization cost, and circuit depth. The variational energy of an ansatz is therefore not always a reliable indicator of its performance when used inside ph-AFQMC.
What carries the argument
Parameterized quantum circuits used as trial wavefunctions inside the phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC) projection, with direct comparison of unitary coupled-cluster (UCCSD), adaptive (ADAPT-VQE), Hamiltonian-informed, and Jastrow-inspired families.
If this is right
- Several ansatz families produce chemically accurate ph-AFQMC energies for hydrogen chains across the entire dissociation curve.
- The variational energy of a trial wavefunction is not a reliable predictor of its quality inside ph-AFQMC.
- At similar parameter counts, different ansatz families give comparable projected energies even when their circuit depths and classical optimization costs differ markedly.
- Over-parameterization occurs in some cases where extra variational parameters do not improve the final ph-AFQMC energy.
Where Pith is reading between the lines
- The findings point to adaptive construction as a practical lever for reducing the quantum circuit resources required to reach a target accuracy in hybrid ph-AFQMC calculations.
- The same benchmarking logic could be applied to larger basis sets or to molecules with more electrons to test whether the resource savings persist.
- The observation that variational energy alone is insufficient suggests that direct optimization of the ph-AFQMC energy itself, rather than the variational energy, may be a useful training objective for future ansatz design.
Load-bearing premise
The advantages seen with adaptive ansatze on linear hydrogen chains under bond stretching will generalize to other strongly correlated molecular systems and are not artifacts of the chosen model or parameter counts.
What would settle it
A calculation on a different strongly correlated molecule, such as stretched water or a small transition-metal complex, in which a fixed-ansatz trial wavefunction yields equal or lower ph-AFQMC energy error than an adaptive ansatz at comparable circuit depth.
Figures



read the original abstract
The phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC) method is a stochastic imaginary-time projection technique for computing ground-state properties of strongly correlated quantum systems, with accuracy that depends critically on the choice of trial wavefunction. Here, we investigate ph-AFQMC with trial states prepared using parameterized quantum circuits. In this work, we present a comprehensive benchmarking study of quantum trial wavefunctions spanning unitary coupled-cluster, Hamiltonian-informed, Jastrow-inspired, and adaptively constructed ansatze. The benchmarking evaluates accuracy, expressibility, and scalability of these ansatze within the QC-AFQMC framework. We test these ansatze on linear hydrogen chains under bond stretching and find that several ansatz families produce chemically accurate ph-AFQMC energies across the dissociation curve. We have performed simulations using the CUDA-Q quantum development platform on the GPU partition of the Perlmutter supercomputer. When comparing ansatze at similar numbers of variational parameters, we find that different ansatz families yield comparable ph-AFQMC results despite exhibiting substantially different variational energies, optimization costs, and circuit depths. Our results indicate that the variational energy of an ansatz is not always a reliable indicator of its quality for ph-AFQMC and reveal instances of over-parameterization. In the strongly correlated regime, trial wavefunctions obtained from adaptive ansatze, exemplified here by ADAPT-VQE with the UCCSD operator pool, can outperform their fixed-ansatz counterparts (UCCSD) in terms of projected energies while using substantially more compact circuits, providing a flexible route to optimize quantum resources within the ph-AFQMC framework.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper benchmarks a range of parameterized quantum-circuit ansatze (UCCSD, Hamiltonian-informed, Jastrow-inspired, and adaptive ADAPT-VQE with UCCSD pool) as trial wavefunctions for phaseless auxiliary-field quantum Monte Carlo. On linear hydrogen chains under bond stretching, the authors report that multiple families reach chemically accurate projected energies, that variational energy is not always a reliable predictor of ph-AFQMC quality, and that adaptive ansatze can yield better projected energies than fixed UCCSD while using substantially more compact circuits in the strongly correlated regime.
Significance. If the reported advantages of adaptive ansatze hold beyond the tested systems, the work supplies concrete guidance for resource-efficient trial-state selection in QC-AFQMC and demonstrates that variational energy alone is an incomplete figure of merit. The GPU-accelerated CUDA-Q implementation on Perlmutter adds a practical reproducibility strength.
major comments (2)
- [Abstract / Numerical results] Abstract and numerical-results section: the central claim that ADAPT-VQE trials 'can outperform their fixed-ansatz counterparts (UCCSD) in terms of projected energies while using substantially more compact circuits' rests exclusively on linear H chains under uniform bond stretching. Because this 1D system exhibits correlation primarily along a single axis, the observed superiority may not extend to 3D molecular dissociation or transition-metal complexes; explicit tests on at least one additional system class are required to support the broader implication for the QC-AFQMC framework.
- [Results] Results section: when ansatze are compared at comparable numbers of variational parameters, the manuscript states that different families produce 'comparable ph-AFQMC results' despite differing variational energies and circuit depths. Without tabulated error bars, statistical uncertainties, or a quantitative definition of 'chemically accurate' (e.g., 1 kcal/mol threshold), it is difficult to judge whether the reported outperformance is statistically significant or merely within noise.
minor comments (2)
- [Abstract] Abstract: the sentence describing the CUDA-Q / Perlmutter simulations interrupts the scientific narrative and would be more appropriate in the Methods or Computational Details section.
- [Results] The manuscript would benefit from a summary table listing each ansatz family, typical parameter count, circuit depth, variational energy, and ph-AFQMC error for the stretched H-chain geometries.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below and have revised the manuscript to incorporate clarifications, additional data presentation, and expanded discussion where feasible.
read point-by-point responses
-
Referee: The central claim that ADAPT-VQE trials 'can outperform their fixed-ansatz counterparts (UCCSD) in terms of projected energies while using substantially more compact circuits' rests exclusively on linear H chains under uniform bond stretching. Because this 1D system exhibits correlation primarily along a single axis, the observed superiority may not extend to 3D molecular dissociation or transition-metal complexes; explicit tests on at least one additional system class are required to support the broader implication for the QC-AFQMC framework.
Authors: We agree that our numerical demonstrations are confined to linear hydrogen chains, a standard benchmark system for strong correlation. The manuscript does not claim universality beyond this class; the reported outperformance and circuit compactness advantages are presented as observations within this well-studied 1D dissociation scenario. To address the concern, we have added a dedicated paragraph in the Conclusions section acknowledging the limitation to 1D chains and explicitly stating that generalization to 3D molecules or transition-metal systems remains an open question requiring future work. We have also softened the abstract language to emphasize that the results provide guidance for the QC-AFQMC framework based on this benchmark rather than a universal claim. No new calculations on additional systems were performed, as they fall outside the current scope. revision: partial
-
Referee: When ansatze are compared at comparable numbers of variational parameters, the manuscript states that different families produce 'comparable ph-AFQMC results' despite differing variational energies and circuit depths. Without tabulated error bars, statistical uncertainties, or a quantitative definition of 'chemically accurate' (e.g., 1 kcal/mol threshold), it is difficult to judge whether the reported outperformance is statistically significant or merely within noise.
Authors: We thank the referee for pointing out the need for clearer statistical presentation. In the revised manuscript we have (i) added a quantitative definition of chemical accuracy as an absolute error below 1 kcal/mol (~1.6 mHa) relative to the reference energy, (ii) included Monte Carlo statistical error bars on all ph-AFQMC energy plots and in a new summary table, and (iii) rephrased the 'comparable results' statement to note that differences lie within the reported uncertainties except in the strongly stretched regime, where the adaptive ansatz advantage exceeds both the error bars and the chemical-accuracy threshold. These changes allow readers to assess significance directly. revision: yes
Circularity Check
No significant circularity detected in empirical benchmarking
full rationale
The paper is a computational benchmarking study that evaluates multiple independent ansatze (UCCSD, ADAPT-VQE, Jastrow-inspired, etc.) by running ph-AFQMC simulations on linear H chains and directly comparing projected energies, circuit depths, and parameter counts. No derivation chain exists; results are obtained from explicit GPU simulations rather than from any fitted parameter renamed as a prediction or from a self-referential definition. The abstract and described methodology contain no load-bearing self-citations, uniqueness theorems, or ansatze smuggled via prior work that would reduce the central claim to its own inputs. The observed outperformance of adaptive ansatze is presented as an empirical finding on the tested systems, not as a mathematically forced consequence.
Axiom & Free-Parameter Ledger
axioms (1)
- standard math Standard quantum mechanics and variational principle for trial wavefunctions
Lean theorems connected to this paper
-
Cost/FunctionalEquation.lean (J=½(x+x⁻¹)-1 uniqueness)washburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
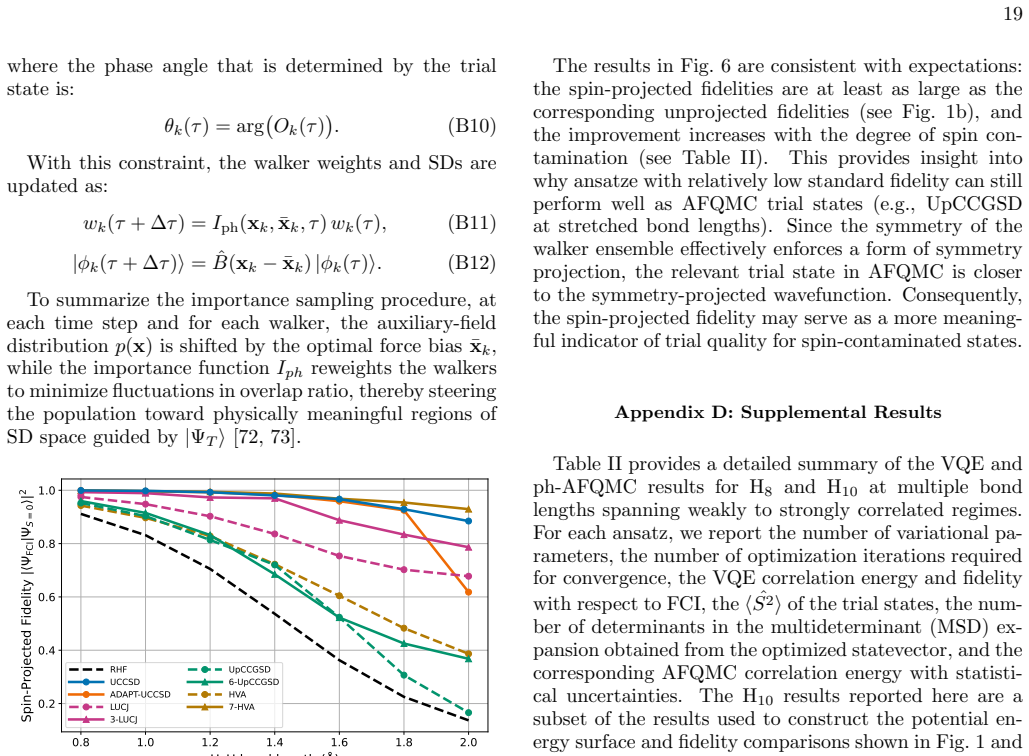
the imaginary-time propagator can be written... ∫ dx p(x) B̂(x) + O(Δτ²)... a Trotter–Suzuki decomposition... a Hubbard–Stratonovich transformation that introduces auxiliary fields.
-
IndisputableMonolith (parameter-free chain)reality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Optimizing the variational parameters that define the trial state... the trial state is prepared on a quantum computer and subsequently used within ph-AFQMC simulations.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
J. Lee, H. Q. Pham, and D. R. Reichman, Twenty years of auxiliary-field quantum monte carlo in quantum chem- istry: An overview and assessment on main group chem- istry and bond-breaking, Journal of Chemical Theory and Computation18, 7024 (2022)
2022
-
[2]
Sugiyama and S
G. Sugiyama and S. Koonin, Auxiliary field monte-carlo for quantum many-body ground states, Annals of Physics 168, 1 (1986)
1986
-
[3]
Pan and Z
G. Pan and Z. Y. Meng, The sign problem in quan- tum monte carlo simulations, inEncyclopedia of Con- densed Matter Physics (Second Edition), edited by T. Chakraborty (Academic Press, Oxford, 2024) second edition ed., pp. 879–893
2024
-
[4]
Zhang, J
S. Zhang, J. Carlson, and J. E. Gubernatis, Constrained path quantum monte carlo method for fermion ground states, Physical review letters74, 3652 (1995)
1995
-
[5]
Zhang and H
S. Zhang and H. Krakauer, Quantum monte carlo method using phase-free random walks with slater determinants, Physical review letters90, 136401 (2003)
2003
-
[6]
M. A. Morales, J. McMinis, B. K. Clark, J. Kim, and G. E. Scuseria, Multideterminant wave functions in quan- tum monte carlo, Journal of chemical theory and compu- tation8, 2181 (2012)
2012
-
[7]
B. K. Clark, M. A. Morales, J. McMinis, J. Kim, and G. E. Scuseria, Computing the energy of a water molecule using multideterminants: A simple, efficient algorithm, The Journal of chemical physics135(2011)
2011
-
[8]
Giner, A
E. Giner, A. Scemama, and M. Caffarel, Using pertur- batively selected configuration interaction in quantum monte carlo calculations, Canadian Journal of Chemistry 91, 879 (2013)
2013
-
[9]
Mahajan and S
A. Mahajan and S. Sharma, Taming the sign problem in auxiliary-field quantum monte carlo using accurate wave functions, Journal of Chemical Theory and Computation 17, 4786 (2021)
2021
-
[10]
B. O. Roos, P. R. Taylor, and P. E. Sigbahn, A complete active space scf method (casscf) using a density matrix formulated super-ci approach, Chemical Physics48, 157 (1980)
1980
-
[11]
E. J. Landinez Borda, J. Gomez, and M. A. Morales, Non-orthogonal multi-slater determinant expansions in auxiliary field quantum monte carlo, The Journal of 14 chemical physics150(2019)
2019
-
[12]
Z.-Y. Xiao, T. Xiang, Z. Lu, Y. Chen, and S. Zhang, Implementing advanced trial wave functions in fermion quantum monte carlo via stochastic sampling, The Jour- nal of Chemical Physics163(2025)
2025
-
[13]
Mahajan and S
A. Mahajan and S. Sharma, Efficient local energy eval- uation for multi-slater wave functions in orbital space quantum monte carlo, The Journal of Chemical Physics 153(2020)
2020
-
[14]
Jiang, B
T. Jiang, B. O’Gorman, A. Mahajan, and J. Lee, Unbi- asing fermionic auxiliary-field quantum monte carlo with matrix product state trial wavefunctions, Physical Re- view Research7, 013038 (2025)
2025
- [15]
-
[16]
W. J. Huggins, B. A. O’Gorman, N. C. Rubin, D. R. Reichman, R. Babbush, and J. Lee, Unbiasing fermionic quantum monte carlo with a quantum computer, Nature 603, 416 (2022)
2022
-
[17]
Huang, R
H.-Y. Huang, R. Kueng, and J. Preskill, Predicting many properties of a quantum system from very few measure- ments, Nature Physics16, 1050 (2020)
2020
-
[18]
G. Mazzola and G. Carleo, Exponential challenges in un- biasing quantum monte carlo algorithms with quantum computers, arXiv preprint arXiv:2205.09203 (2022)
- [19]
-
[20]
K. Wan, W. J. Huggins, J. Lee, and R. Babbush, Match- gate shadows for fermionic quantum simulation, Commu- nications in Mathematical Physics404, 629 (2023)
2023
-
[21]
Huang, Y.-T
B. Huang, Y.-T. Chen, B. Gupt, M. Suchara, A. Tran, S. McArdle, and G. Galli, Evaluating a quantum-classical quantum monte carlo algorithm with matchgate shadows, Physical Review Research6, 043063 (2024)
2024
-
[22]
Kiser, A
M. Kiser, A. Schroeder, G.-L. R. Anselmetti, C. Ku- mar, N. Moll, M. Streif, and D. Vodola, Classical and quantum cost of measurement strategies for quantum- enhanced auxiliary field quantum monte carlo, New Jour- nal of Physics26, 033022 (2024)
2024
-
[23]
L. Zhao, J. J. Goings, W. Aboumrad, A. Arrasmith, L. Calderin, S. Churchill, D. Gabay, T. Harvey-Brown, M. Hiles, M. Kaja,et al., Quantum-classical auxil- iary field quantum monte carlo with matchgate shad- ows on trapped ion quantum computers, arXiv preprint arXiv:2506.22408 (2025)
-
[24]
Amsler, P
M. Amsler, P. Deglmann, M. Degroote, M. P. Kaicher, M. Kiser, M. K¨ uhn, C. Kumar, A. Maier, G. Samsonidze, A. Schroeder,et al., Classical and quantum trial wave functions in auxiliary-field quantum monte carlo applied to oxygen allotropes and a cubr2 model system, The Journal of Chemical Physics159(2023)
2023
-
[25]
V. Khinevich and W. Mizukami, Enhancing quantum computations with the synergy of auxiliary field quan- tum monte carlo and computational basis tomography, arXiv preprint arXiv:2502.20066 (2025)
- [26]
-
[27]
J. J. Goings, K. Shin, S. Noh, W. Kyoung, D. Kim, J. Baek, M. Roetteler, E. Epifanovsky, and L. Zhao, Molecular properties in quantum-classical auxiliary-field quantum monte carlo: Correlated sampling with ap- plication to accurate nuclear forces, arXiv preprint arXiv:2507.17992 (2025)
-
[28]
N. S. Blunt, L. Caune, and J. Quiroz-Fernandez, Quan- tum computing approach to fixed-node monte carlo using classical shadows, Journal of Chemical Theory and Com- putation21, 1652 (2025)
2025
-
[29]
Kiser, M
M. Kiser, M. Beuerle, and F. Simkovic IV, Contextual subspace auxiliary-field quantum monte carlo: Improved bias with reduced quantum resources, Journal of Chem- ical Theory and Computation21, 2256 (2025)
2025
-
[30]
Y. Yoshida, L. Erhart, T. Murokoshi, R. Nakagawa, C. Mori, T. Miyanaga, T. Mori, and W. Mizukami, Auxiliary-field quantum monte carlo method with quan- tum selected configuration interaction, arXiv preprint arXiv:2502.21081 (2025)
-
[31]
Motta, D
M. Motta, D. M. Ceperley, G. K.-L. Chan, J. A. Gomez, E. Gull, S. Guo, C. A. Jim´ enez-Hoyos, T. N. Lan, J. Li, F. Ma,et al., Towards the solution of the many-electron problem in real materials: Equation of state of the hy- drogen chain with state-of-the-art many-body methods, Physical Review X7, 031059 (2017)
2017
-
[32]
Hachmann, W
J. Hachmann, W. Cardoen, and G. K. Chan, Multiref- erence correlation in long molecules with the quadratic scaling density matrix renormalization group, The Jour- nal of chemical physics125(2006)
2006
-
[33]
W. J. Huggins, J. R. McClean, N. C. Rubin, Z. Jiang, N. Wiebe, K. B. Whaley, and R. Babbush, Efficient and noise resilient measurements for quantum chemistry on near-term quantum computers, npj Quantum Informa- tion7, 23 (2021)
2021
-
[34]
If you use this software, please cite it as in- structed
The CUDA-Q development team, Cuda-q,https:// github.com/NVIDIA/cuda-quantum(2024), apache-2.0 License. If you use this software, please cite it as in- structed
2024
-
[35]
L. Zhao, J. Goings, K. Shin, W. Kyoung, J. I. Fuks, J.-K. Kevin Rhee, Y. M. Rhee, K. Wright, J. Nguyen, J. Kim, et al., Orbital-optimized pair-correlated electron simula- tions on trapped-ion quantum computers, npj Quantum Information9, 60 (2023)
2023
-
[36]
Purwanto, W
W. Purwanto, W. Al-Saidi, H. Krakauer, and S. Zhang, Eliminating spin contamination in auxiliary-field quan- tum monte carlo: Realistic potential energy curve of f2, The Journal of chemical physics128(2008)
2008
-
[37]
J. Lee, F. D. Malone, and M. A. Morales, An auxiliary- field quantum monte carlo perspective on the ground state of the dense uniform electron gas: An investiga- tion with hartree-fock trial wavefunctions, The Journal of Chemical Physics151(2019)
2019
-
[38]
W. A. Al-Saidi, S. Zhang, and H. Krakauer, Bond break- ing with auxiliary-field quantum monte carlo, The Jour- nal of chemical physics127(2007)
2007
-
[39]
Quantum Computation by Adiabatic Evolution
E. Farhi, J. Goldstone, S. Gutmann, and M. Sipser, Quantum computation by adiabatic evolution, arXiv preprint quant-ph/0001106 (2000)
work page Pith review arXiv 2000
-
[40]
Aspuru-Guzik, A
A. Aspuru-Guzik, A. D. Dutoi, P. J. Love, and M. Head- Gordon, Simulated quantum computation of molecular energies, Science309, 1704 (2005)
2005
-
[41]
D. S. Abrams and S. Lloyd, Quantum algorithm provid- ing exponential speed increase for finding eigenvalues and 15 eigenvectors, Physical Review Letters83, 5162 (1999)
1999
-
[42]
Jordan and E
P. Jordan and E. Wigner, ¨Uber das paulische ¨ aquivalenzverbot, Zeitschrift f¨ ur Physik47, 631 (1928)
1928
-
[43]
S. B. Bravyi and A. Y. Kitaev, Fermionic quantum com- putation, Annals of Physics298, 210 (2002)
2002
-
[44]
J. R. McClean, S. Boixo, V. N. Smelyanskiy, R. Bab- bush, and H. Neven, Barren plateaus in quantum neural network training landscapes, Nature communications9, 4812 (2018)
2018
-
[45]
Cerezo, A
M. Cerezo, A. Arrasmith, R. Babbush, S. C. Benjamin, S. Endo, K. Fujii, J. R. McClean, K. Mitarai, X. Yuan, L. Cincio,et al., Variational quantum algorithms, Nature Reviews Physics3, 625 (2021)
2021
-
[46]
Peruzzo, J
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’brien, A variational eigenvalue solver on a photonic quantum processor, Nature communications5, 4213 (2014)
2014
-
[47]
Møller and M
C. Møller and M. S. Plesset, Note on an approximation treatment for many-electron systems, Physical review46, 618 (1934)
1934
-
[48]
Romero, R
J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love, and A. Aspuru-Guzik, Strategies for quantum com- puting molecular energies using the unitary coupled clus- ter ansatz, Quantum Science and Technology4, 014008 (2018)
2018
-
[49]
M. R. Hirsbrunner, D. Chamaki, J. W. Mullinax, and N. M. Tubman, Beyond mp2 initialization for unitary coupled cluster quantum circuits, Quantum8, 1538 (2024)
2024
-
[50]
Wecker, M
D. Wecker, M. B. Hastings, and M. Troyer, Progress to- wards practical quantum variational algorithms, Physical Review A92, 042303 (2015)
2015
-
[51]
Park and N
C.-Y. Park and N. Killoran, Hamiltonian variational ansatz without barren plateaus, Quantum8, 1239 (2024)
2024
-
[52]
Wiersema, C
R. Wiersema, C. Zhou, Y. de Sereville, J. F. Carrasquilla, Y. B. Kim, and H. Yuen, Exploring entanglement and optimization within the hamiltonian variational ansatz, PRX quantum1, 020319 (2020)
2020
-
[53]
Matsuzawa and Y
Y. Matsuzawa and Y. Kurashige, Jastrow-type decom- position in quantum chemistry for low-depth quantum circuits, Journal of chemical theory and computation16, 944 (2020)
2020
- [54]
-
[55]
J. Lee, W. J. Huggins, M. Head-Gordon, and K. B. Wha- ley, Generalized unitary coupled cluster wave functions for quantum computation, Journal of chemical theory and computation15, 311 (2018)
2018
-
[56]
Motta, K
M. Motta, K. J. Sung, K. B. Whaley, M. Head-Gordon, and J. Shee, Bridging physical intuition and hardware ef- ficiency for correlated electronic states: the local unitary cluster jastrow ansatz for electronic structure, Chemical Science14, 11213 (2023)
2023
-
[57]
Motta, K
M. Motta, K. J. Sung, and J. Shee, Quantum algo- rithms for the variational optimization of correlated elec- tronic states with stochastic reconfiguration and the lin- ear method, The Journal of Physical Chemistry A128, 8762 (2024)
2024
-
[58]
H. R. Grimsley, S. E. Economou, E. Barnes, and N. J. Mayhall, An adaptive variational algorithm for exact molecular simulations on a quantum computer, Nature communications10, 3007 (2019)
2019
-
[59]
P. K. Barkoutsos, J. F. Gonthier, I. Sokolov, N. Moll, G. Salis, A. Fuhrer, M. Ganzhorn, D. J. Egger, M. Troyer, A. Mezzacapo,et al., Quantum algorithms for electronic structure calculations: Particle-hole hamiltonian and op- timized wave-function expansions, Physical Review A98, 022322 (2018)
2018
-
[60]
Chen, H.-P
J. Chen, H.-P. Cheng, and J. K. Freericks, Quantum- inspired algorithm for the factorized form of unitary cou- pled cluster theory, Journal of Chemical Theory and Computation17, 841 (2021)
2021
-
[61]
Wecker, M
D. Wecker, M. B. Hastings, N. Wiebe, B. K. Clark, C. Nayak, and M. Troyer, Solving strongly correlated electron models on a quantum computer, Phys. Rev. A 92, 062318 (2015)
2015
-
[62]
Jiang, K
Z. Jiang, K. J. Sung, K. Kechedzhi, V. N. Smelyanskiy, and S. Boixo, Quantum algorithms to simulate many- body physics of correlated fermions, Physical Review Ap- plied9, 044036 (2018)
2018
- [63]
-
[64]
The ffsim developers, ffsim: Faster simulations of fermionic quantum circuits,https://github.com/ qiskit-community/ffsim
-
[65]
Carlson, J
J. Carlson, J. Gubernatis, G. Ortiz, and S. Zhang, Issues and observations on applications of the constrained-path monte carlo method to many-fermion systems, Physical Review B59, 12788 (1999)
1999
-
[66]
Mahajan, J
A. Mahajan, J. H. Thorpe, J. S. Kurian, D. R. Reichman, D. A. Matthews, and S. Sharma, Beyond ccsd (t) accu- racy at lower scaling with auxiliary field quantum monte carlo, Journal of Chemical Theory and Computation21, 1626 (2025)
2025
-
[67]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui, et al., Recent developments in the pyscf program package, The Journal of chemical physics153(2020)
2020
-
[68]
Virtanen, R
P. Virtanen, R. Gommers, T. E. Oliphant, M. Haber- land, T. Reddy, D. Cournapeau, E. Burovski, P. Pe- terson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, ˙I. Po- lat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henr...
2020
-
[69]
J. R. McClean, N. C. Rubin, K. J. Sung, I. D. Kivlichan, X. Bonet-Monroig, Y. Cao, C. Dai, E. S. Fried, C. Gid- ney, B. Gimby,et al., Openfermion: the electronic struc- ture package for quantum computers, Quantum Science and Technology5, 034014 (2020)
2020
-
[70]
H. R. Grimsley, D. Claudino, S. E. Economou, E. Barnes, and N. J. Mayhall, Is the trotterized uccsd ansatz chem- ically well-defined?, Journal of chemical theory and com- putation16, 1 (2019)
2019
-
[71]
A. Javadi-Abhari, M. Treinish, K. Krsulich, C. J. Wood, J. Lishman, J. Gacon, S. Martiel, P. D. Nation, L. S. Bishop, A. W. Cross, B. R. Johnson, and J. M. Gambetta, Quantum computing with Qiskit (2024), 16 arXiv:2405.08810 [quant-ph]
work page internal anchor Pith review arXiv 2024
-
[72]
F. D. Malone, A. Mahajan, J. S. Spencer, and J. Lee, Ipie: A python-based auxiliary-field quantum monte carlo pro- gram with flexibility and efficiency on cpus and gpus, Journal of Chemical Theory and Computation19, 109 (2022)
2022
-
[73]
Pavarini, E
E. Pavarini, E. Koch, and U. Schollw¨ ock,Emer- gent Phenomena in Correlated Matter: Lecture Notes of the Autumn School Correlated Electrons 2013, at Forschungszentrum J¨ ulich, 23-27 September 2013, Vol. 3 (Forschungszentrum J¨ ulich, 2013). Appendix A: Simulation Details All Hamiltonian integrals, SCF, CCSD, and FCI cal- culations were performed using t...
2013
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.