Recognition: 1 theorem link
· Lean TheoremFrom Enhanced Sampling to Human-Readable Representations of Protein Dynamics
Pith reviewed 2026-05-11 00:44 UTC · model grok-4.3
The pith
An automated workflow converts complex collective variables into simple protein domain distances.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The framework transforms enhanced sampling trajectories into human-readable protein dynamics representations. Weighted dynamic cross-correlation matrices applied post hoc identify correlated residue pairs and domains, producing simple domain-domain distances as interpretable collective variables. For five proteins including KRAS and HIV-1 protease, free energy surfaces from these distances reproduce known conformational states with low uncertainty while maximizing independent dynamical information.
What carries the argument
Weighted dynamic cross-correlation matrices applied post hoc to biased trajectories to identify correlated domains and derive simple geometric distances as new collective variables.
If this is right
- Free energy surfaces from the distances reproduce known states for tested proteins.
- The approach works without system-specific knowledge for multiple proteins.
- Complex CVs are recast into geometric forms without losing essential dynamics.
- Resulting ensembles support integration with machine learning.
- It offers a general tool for interpreting enhanced sampling of protein dynamics.
Where Pith is reading between the lines
- The workflow could generate standardized interpretable data sets from various enhanced sampling protocols.
- Human-readable representations may aid hypothesis generation on how domain motions relate to protein function.
- It provides a path to combine physics-based sampling with data-driven methods for conformational analysis.
Load-bearing premise
The method assumes that weighted dynamic cross-correlation matrices calculated from biased trajectories will extract biologically meaningful domains and motions without artifacts from the sampling bias.
What would settle it
If the domain-domain distances fail to produce free energy surfaces that match known conformational states for HIV-1 protease when tested against reference data, the central claim would be falsified.
Figures




read the original abstract
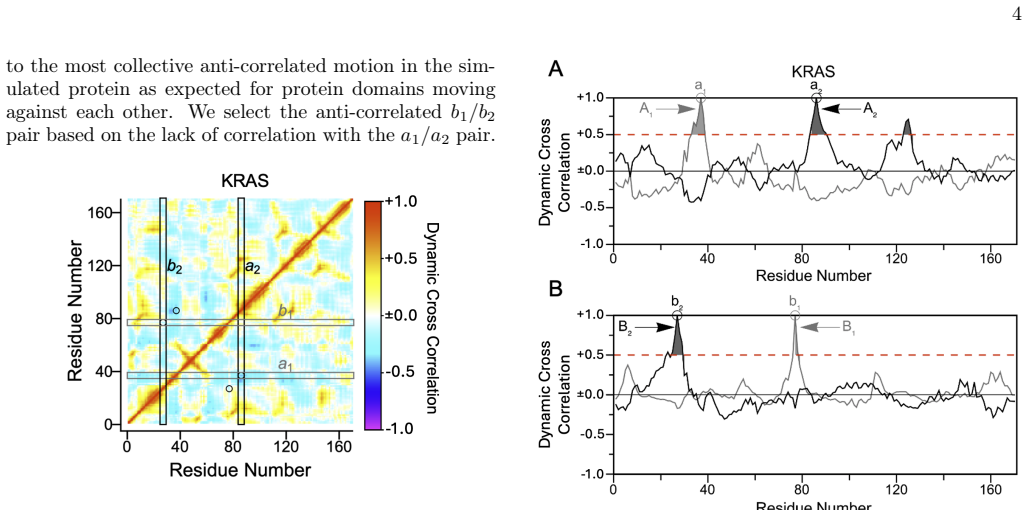
Understanding protein conformational dynamics is essential for elucidating biological function but remains challenging due to the wide range of timescales and the complexity of collective motions. Enhanced sampling methods overcome timescale limitations of conventional molecular dynamics, yet their effectiveness depends on the choice of collective variables (CVs), which are often difficult to define and may lack physical interpretability. In particular, collective variables derived from machine learning or collective vibrational modes can efficiently capture slow dynamics but are not easily mapped onto intuitive structural descriptors. Here, we present a fully automated framework that transforms enhanced sampling trajectories into human-readable representations of protein dynamics. Our approach combines enhanced sampling along CVs derived from frequency-selective anharmonic mode analysis with a post hoc analysis of biased trajectories using weighted dynamic cross-correlation matrices. From these, we identify residue pairs and domains exhibiting correlated and anti-correlated motions, yielding simple domain-domain distances that serve as physically interpretable CVs. We apply this method to five proteins, including KRAS and HIV-1 protease, and show that it consistently identifies biologically relevant domains and motions without prior system-specific knowledge. Projection onto these distances produces free energy surfaces that reproduce known conformational states with low statistical uncertainty while maximizing independent dynamical information. This workflow enables systematic recasting of complex CVs into simple geometric descriptors without loss of essential dynamics. Its generality and automation make it broadly applicable for interpreting enhanced sampling simulations and generating interpretable conformational ensembles for integration with emerging machine learning approaches.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a fully automated framework that combines enhanced sampling along collective variables derived from frequency-selective anharmonic mode analysis with post-hoc analysis of the biased trajectories using weighted dynamic cross-correlation matrices (DCCM). From the resulting correlated and anti-correlated residue motions, the method identifies domains and derives simple domain-domain distances as new, physically interpretable collective variables. Applied to five proteins (including KRAS and HIV-1 protease) without prior system-specific knowledge, the approach yields free energy surfaces that reproduce known conformational states with low statistical uncertainty while maximizing independent dynamical information. The central claim is that this workflow systematically recasts complex CVs into human-readable geometric descriptors without loss of essential dynamics.
Significance. If the central claim holds, the work would be significant for bridging machine-learning or mode-derived CVs with intuitive structural descriptors in molecular biophysics. The automation, lack of system-specific tuning, and demonstration across multiple proteins (with explicit credit for reproducible application to KRAS and HIV-1 protease) are strengths that could aid interpretation of enhanced sampling results and integration with emerging ML-based ensemble methods. The emphasis on maximizing independent dynamical information and producing falsifiable geometric CVs adds value if the post-hoc reweighting is robust.
major comments (3)
- [§3.2] §3.2 (Weighted DCCM procedure): The reweighting of dynamic cross-correlation matrices to recover equilibrium fluctuations from biased trajectories is load-bearing for the claim of no loss of essential dynamics, yet the manuscript provides no explicit convergence diagnostics, comparison to reference unbiased simulations, or tests for residual artifacts from history-dependent bias or incomplete orthogonal-mode sampling. This directly addresses the skeptic concern and must be strengthened to support the generality assertion.
- [Results section] Results (application to five proteins): The abstract and results state that the derived distances reproduce known conformational states with low statistical uncertainty, but the quantitative error analysis (e.g., bootstrap uncertainties, overlap metrics with reference distributions, or sensitivity to post-hoc domain selection) is not detailed sufficiently to confirm the claim across all systems; without this, the 'low uncertainty' and 'biologically relevant' assertions remain under-supported.
- [§4.1] §4.1 (Free energy surfaces): The projection onto the new domain-domain distances is said to maximize independent dynamical information, but no quantitative measure (e.g., mutual information or principal-component overlap with the original anharmonic modes) is provided to demonstrate preservation versus loss of essential dynamics, which is central to the workflow's validity.
minor comments (3)
- [Figure 3] Figure 3 (domain visualizations): The residue-pair correlation maps would benefit from explicit scale bars and annotation of the identified domain boundaries to improve readability.
- [Introduction] Introduction: A brief comparison table or paragraph contrasting the new workflow with prior DCCM-based or mode-analysis methods would clarify the incremental advance.
- [Methods] Notation: The definition of 'weighted DCCM' could be formalized as an equation (currently described in prose) to aid reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed report. The comments identify areas where additional validation and quantification would strengthen the manuscript, and we address each point below with plans for revision.
read point-by-point responses
-
Referee: [§3.2] §3.2 (Weighted DCCM procedure): The reweighting of dynamic cross-correlation matrices to recover equilibrium fluctuations from biased trajectories is load-bearing for the claim of no loss of essential dynamics, yet the manuscript provides no explicit convergence diagnostics, comparison to reference unbiased simulations, or tests for residual artifacts from history-dependent bias or incomplete orthogonal-mode sampling. This directly addresses the skeptic concern and must be strengthened to support the generality assertion.
Authors: We agree that the weighted DCCM reweighting requires stronger validation to support the no-loss claim. In the revised manuscript we will add convergence diagnostics showing stabilization of key DCCM elements with increasing trajectory length for all five systems. We will also report tests for residual bias artifacts by comparing weighted DCCM results obtained under different bias strengths and orthogonal-mode sampling conditions. Where literature unbiased trajectories exist (HIV-1 protease and KRAS), we will include direct comparisons of the recovered correlations. revision: yes
-
Referee: [Results section] Results (application to five proteins): The abstract and results state that the derived distances reproduce known conformational states with low statistical uncertainty, but the quantitative error analysis (e.g., bootstrap uncertainties, overlap metrics with reference distributions, or sensitivity to post-hoc domain selection) is not detailed sufficiently to confirm the claim across all systems; without this, the 'low uncertainty' and 'biologically relevant' assertions remain under-supported.
Authors: We acknowledge that the current quantitative support for low uncertainty and biological relevance is insufficient. The revised Results section will include bootstrap-derived uncertainties on the free-energy surfaces and domain distances for every protein. We will also add overlap metrics (Jensen-Shannon divergence) against reference conformational distributions and a brief sensitivity analysis to the automated domain-selection thresholds. revision: yes
-
Referee: [§4.1] §4.1 (Free energy surfaces): The projection onto the new domain-domain distances is said to maximize independent dynamical information, but no quantitative measure (e.g., mutual information or principal-component overlap with the original anharmonic modes) is provided to demonstrate preservation versus loss of essential dynamics, which is central to the workflow's validity.
Authors: We agree that a quantitative demonstration of dynamical-information preservation is needed. In the revised §4.1 we will report mutual information between the original frequency-selective anharmonic modes and the final domain-domain distances, together with the principal-component overlap between the dynamics captured by the new CVs and the original modes. revision: yes
Circularity Check
No significant circularity; workflow derives new CVs via post-hoc analysis without reduction to inputs by construction
full rationale
The claimed derivation applies frequency-selective anharmonic mode analysis for initial enhanced sampling, followed by independent post-hoc weighted DCCM computation on the resulting trajectories to extract correlated residue pairs and domain distances. These steps produce new geometric descriptors that are then validated by reproducing known conformational states on five proteins, providing external grounding rather than tautological equivalence. No equations or steps reduce the output CVs to the input CVs by definition, no self-citation chains bear the central claim, and no fitted parameters are relabeled as predictions. The pipeline remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearOur approach combines enhanced sampling along CVs derived from frequency-selective anharmonic mode analysis with a post hoc analysis of biased trajectories using weighted dynamic cross-correlation matrices... yielding simple domain-domain distances
Reference graph
Works this paper leans on
-
[1]
Henzler-Wildman and D
K. Henzler-Wildman and D. Kern, Dynamic personalities of proteins, Nature450, 964 (2007)
2007
-
[2]
W.J.Wedemeyer, E.Welker,andH.A.Scheraga,Proline cis- transisomerization andprotein folding,Biochemistry 8 41, 14637 (2002)
2002
-
[3]
D. D. Boehr, H. J. Dyson, and P. E. Wright, An nmr perspective on enzyme dynamics, Chem. Rev.106, 3055 (2006)
2006
-
[4]
Karplus and J
M. Karplus and J. A. McCammon, Molecular dynam- ics simulations of biomolecules, Nat. Struct. Biol.9, 646 (2002)
2002
-
[5]
R. O. Dror, R. M. Dirks, J. Grossman, H. Xu, and D. E. Shaw, Biomolecular simulation: A computational micro- scope for molecular biology, Annu. Rev. Biophys.41, 429 (2012)
2012
-
[6]
D. E. Shaw, P. J. Adams, A. Azaria, J. A. Bank, B. Bat- son, A. Bell, M. Bergdorf, J. Bhatt, J. A. Butts, T. Cor- reia,et al., Anton 3: Twenty microseconds of molecular dynamics simulation before lunch, inProceedings of the International Conference for High Performance Comput- ing, Networking, Storage and Analysis(2021) pp. 1–11
2021
-
[7]
P. Ayaz, A. Lyczek, Y. Paung, V. R. Mingione, R. E. Ia- cob, P. W. de Waal, J. R. Engen, M. A. Seeliger, Y. Shan, and D. E. Shaw, Structural mechanism of a drug-binding process involving a large conformational change of the protein target, Nat. Commun.14, 1885 (2023)
2023
-
[8]
J. B. Greisman, L. Willmore, C. Y. Yeh, F. Giordanetto, S. Shahamadtar, H. Nisonoff, P. Maragakis, and D. E. Shaw, Discovery and validation of the binding poses of allosteric fragment hits to protein tyrosine phosphatase 1b: From molecular dynamics simulations to x-ray crys- tallography, J. Chem. Inf. and Model.63, 2644 (2023)
2023
-
[9]
Lewis, T
S. Lewis, T. Hempel, J. Jiménez-Luna, M. Gastegger, Y. Xie, A. Y. Foong, V. G. Satorras, O. Abdin, B. S. Veeling, I. Zaporozhets, Y. Chen, S. Yang, A. E. Foster, A. Schneuing, J. Nigam, F. Barbero, V. Stimper, A. Campbell, J. Yim, M. Lienen, Y. Shi, S. Zheng, H. Schulz, U. Munir, R. Sordillo, R. Tomioka, C. Clementi, and F. Noé, Scalable emulation of prot...
2025
-
[10]
R. C. Bernardi, M. C. Melo, and K. Schulten, Enhanced sampling techniques in molecular dynamics simulations of biological systems, Biochim. Biophys. Acta, Gen. Subj. 1850, 872 (2015)
2015
-
[11]
Mitsutake, Y
A. Mitsutake, Y. Sugita, and Y. Okamoto, Generalized- ensemblealgorithmsformolecularsimulationsofbiopoly- mers, Biomolecules60, 96 (2001)
2001
-
[12]
G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in monte carlo free-energy estimation: Um- brella sampling, J. Comput. Phys.23, 187 (1977)
1977
-
[13]
Isralewitz, J
B. Isralewitz, J. Baudry, J. Gullingsrud, D. Kosztin, and K. Schulten, Steered molecular dynamics investigations of protein function, J. Mol. Graph. Model.19, 13 (2001)
2001
-
[14]
Schlitter, M
J. Schlitter, M. Engels, and P. Krüger, Targeted molecu- lar dynamics: a new approach for searching pathways of conformational transitions, J. Mol. Graph.12, 84 (1994)
1994
-
[15]
Hénin and C
J. Hénin and C. Chipot, Overcoming free energy barriers using unconstrained molecular dynamics simulations, J. Chem. Phys.121, 2904 (2004)
2004
-
[16]
Laio and M
A. Laio and M. Parrinello, Escaping free-energy minima, Proc. Natl. Acad. Sci. U.S.A99, 12562 (2002)
2002
-
[17]
Sugita and Y
Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett. 314, 141 (1999)
1999
-
[18]
Hamelberg, J
D. Hamelberg, J. Mongan, and J. A. McCammon, Ac- celerated molecular dynamics: a promising and efficient simulation method for biomolecules, J. Chem. Phys.120, 11919 (2004)
2004
-
[19]
Mehdi, Z
S. Mehdi, Z. Smith, L. Herron, Z. Zou, and P. Tiwary, Enhanced sampling with machine learning, Ann. Rev. Phys. Chem.75(2024)
2024
-
[20]
N. Go, T. Noguti, and T. Nishikawa, Dynamics of a small globular protein in terms of low-frequency vibrational modes., Proc. Natl. Acad. Sci. U.S.A80, 3696 (1983)
1983
-
[21]
D. v. Aalten, J. Findlay, A. Amadei, and H. Berendsen, Essential dynamics of the cellular retinol-binding protein evidence for ligand-induced conformational changes, Pro- tein Eng. Des. Sel.8, 1129 (1995)
1995
-
[22]
Hayward, A
S. Hayward, A. Kitao, and N. G¯ o, Harmonicity and an- harmonicity in protein dynamics: a normal mode anal- ysis and principal component analysis, Proteins. Struct. Funct. Bioinf.23, 177 (1995)
1995
-
[23]
Ma, Usefulness and limitations of normal mode anal- ysis in modeling dynamics of biomolecular complexes, Structure13, 373 (2005)
J. Ma, Usefulness and limitations of normal mode anal- ysis in modeling dynamics of biomolecular complexes, Structure13, 373 (2005)
2005
-
[24]
M. A. Sauer and M. Heyden, Frequency-selective anhar- monic mode analysis of thermally excited vibrations in proteins, J. Chem. Theory Comput.19, 5481 (2023)
2023
-
[25]
A. E. García, Large-amplitude nonlinear motions in pro- teins, Phys. Rev. Lett.68, 2696 (1992)
1992
-
[26]
Amadei, A
A. Amadei, A. B. Linssen, and H. J. Berendsen, Essential dynamics of proteins, Proteins Struct. Funct. Bioinf.17, 412 (1993)
1993
-
[27]
Naritomi and S
Y. Naritomi and S. Fuchigami, Slow dynamics of a pro- tein backbone in molecular dynamics simulation revealed by time-structure based independent component analy- sis, J. Chem. Phys.139(2013)
2013
-
[28]
Noé and C
F. Noé and C. Clementi, Kinetic distance and kinetic maps from molecular dynamics simulation, J. Chem. Theory Comput.11, 5002 (2015)
2015
-
[29]
Sidky, W
H. Sidky, W. Chen, and A. L. Ferguson, Machine learning for collective variable discovery and enhanced sampling in biomolecular simulation, Mol. Phys.118, e1737742 (2020)
2020
-
[30]
Y. Wang, J. M. L. Ribeiro, and P. Tiwary, Machine learn- ing approaches for analyzing and enhancing molecular dynamics simulations, Curr. Opin. Struct. Biol.61, 139 (2020)
2020
-
[31]
F. Noé, A. Tkatchenko, K.-R. Müller, and C. Clementi, Machine learning for molecular simulation, Annu. Rev. Phys. Chem.71, 361 (2020)
2020
-
[32]
Hanni and D
J. Hanni and D. Ray, Data efficient learning of molec- ular slow modes from nonequilibrium metadynamics, J. Chem. Phys.162(2025)
2025
-
[33]
H. Fu, H. Bian, X. Shao, and W. Cai, Collective variable- based enhanced sampling: From human learning to ma- chine learning, J. Phys. Chem. Lett.15, 1774 (2024)
2024
-
[34]
Fröhlking, L
T. Fröhlking, L. Bonati, V. Rizzi, and F. L. Gerva- sio, Deep learning path-like collective variable for en- hanced sampling molecular dynamics, J. Chem. Phys. 160(2024)
2024
-
[35]
Mondal, M
S. Mondal, M. A. Sauer, and M. Heyden, Exploring con- formational landscapes along anharmonic low-frequency vibrations, J. Phys. Chem. B128, 7112 (2024)
2024
-
[36]
M. A. Sauer, S. Mondal, M. Cano, and M. Heyden, High- throughput computation of anharmonic low-frequency protein vibrations, J. Phys. Chem. B129, 10739 (2025)
2025
-
[37]
M. A. Sauer, S. Mondal, B. Neff, S. Maiti, and M. Hey- den, Fast sampling of protein conformational dynamics, Science Advances12, eaea4617 (2026). 9
2026
-
[38]
B. Neff and M. Heyden, Protein-water energy trans- fer via anharmonic low-frequency vibrations (2026), arXiv:2601.02699 [cond-mat.soft]
-
[39]
McCammon, Protein dynamics, Rep
J. McCammon, Protein dynamics, Rep. Prog. Phys.47, 1 (1984)
1984
-
[40]
Hornak, A
V. Hornak, A. Okur, R. C. Rizzo, and C. Simmerling, Hiv-1 protease flaps spontaneously open and reclose in molecular dynamics simulations, Proc. Natl. Acad. Sci. U.S.A103, 915 (2006)
2006
-
[41]
Pantsar, The current understanding of kras protein structure and dynamics, Comput
T. Pantsar, The current understanding of kras protein structure and dynamics, Comput. Struct. Biotechnol. J. 18, 189 (2020)
2020
-
[42]
H. J. Kim, H. N. Lee, M. S. Jeong, and S. B. Jang, Onco- genic kras: signaling and drug resistance, Cancers13, 5599 (2021)
2021
-
[43]
V. Jani, U. Sonavane, and R. Joshi, Insight into struc- tural dynamics involved in activation mechanism of full length kras wild type and p-loop mutants, Heliyon10 (2024)
2024
-
[44]
N. Bery, S. Legg, J. Debreczeni, J. Breed, K. Embrey, C. Stubbs, P. Kolasinska-Zwierz, N. Barrett, R. Mar- wood, J. Watson,et al., Kras-specific inhibition using a darpin binding to a site in the allosteric lobe, Nat. Com- mun.10, 2607 (2019)
2019
-
[45]
M. G. Costa, P. R. Batista, A. Gomes, L. S. Bastos, M. Louet, N. Floquet, P. M. Bisch, and D. Perahia, Mdexciter: Enhanced sampling molecular dynamics by excited normal modes or principal components obtained from experiments, J. Chem. Theory Comput.19, 412 (2023)
2023
-
[46]
Y. M. Huang, J. A. McCammon, and Y. Miao, Replica exchange gaussian accelerated molecular dynamics: Im- proved enhanced sampling and free energy calculation, J. Chem. Theory Comput.14, 1853 (2018)
2018
-
[47]
W. Ren, H. M. Dokainish, A. Shinobu, H. Oshima, and Y. Sugita, Unraveling the coupling between conforma- tional changes and ligand binding in ribose binding pro- tein using multiscale molecular dynamics and free-energy calculations, J. Phys. Chem. B125, 2898 (2021)
2021
-
[48]
Benabderrahmane, R
M. Benabderrahmane, R. Bureau, A. S. Voisin-Chiret, and J. Sopkova-de Oliveira Santos, Insights into mcl- 1 conformational states and allosteric inhibition mech- anism from molecular dynamics simulations, enhanced sampling, and pocket crosstalk analysis, J. Chem. Inf. Model.60, 3172 (2020)
2020
-
[49]
Brooks and M
B. Brooks and M. Karplus, Harmonic dynamics of pro- teins: normal modes and fluctuations in bovine pancre- atic trypsin inhibitor., Proc. Natl. Acad. Sci. U.S.A80, 6571 (1983)
1983
-
[50]
J. M. L. Ribeiro, P. Bravo, Y. Wang, and P. Tiwary, Reweighted autoencoded variational bayes for enhanced sampling (rave), J. Chem. Phys.149(2018)
2018
-
[51]
Y. Wang, J. M. L. Ribeiro, and P. Tiwary, Past–future information bottleneck for sampling molecular reaction coordinate simultaneously with thermodynamics and ki- netics, Nat. Commun.10, 3573 (2019)
2019
-
[52]
Istomin and G
V. Istomin and G. Piccini, Festa: A polygon-based ap- proach for extracting relevant structures from free energy surfaces obtained in molecular simulations, J. Chem. Inf. Model.65, 1 (2024)
2024
-
[53]
Chatterjee and D
S. Chatterjee and D. Ray, Acceleration with inter- pretability: A surrogate model-based collective variable for enhanced sampling, J. Chem. Theory Comput.21, 1561 (2025)
2025
-
[54]
De Simone, R
A. De Simone, R. W. Montalvao, C. M. Dobson, and M. Vendruscolo, Characterization of the interdomain mo- tions in hen lysozyme using residual dipolar couplings as replica-averaged structural restraints in molecular dy- namics simulations, Biochemistry52, 6480 (2013)
2013
-
[55]
J. Chen, S. Zhang, W. Wang, L. Pang, Q. Zhang, and X. Liu, Mutation-induced impacts on the switch trans- formations of the gdp-and gtp-bound k-ras: insights from multiple replica gaussian accelerated molecular dynamics and free energy analysis, J. Chem. Inf. Model.61, 1954 (2021)
1954
-
[56]
Barducci, G
A. Barducci, G. Bussi, and M. Parrinello, Well-tempered metadynamics: a smoothly converging and tunable free- energy method, Phys. Rev. Lett.100, 020603 (2008)
2008
-
[57]
M. A. Sauer, S. Mondal, and M. Heyden, Fresean metadynamics with weighted DCCM analysis DOI: 10.5281/zenodo.20076452 (2026). 1 Supporting Information: From Enhanced Sampling to Human-Readable Representations of Protein Dynamics FIG. S1. Dynamic cross-correlation matrices (DCCMs) computed as weighted averages from enhanced sampling simulations for HEWL, HI...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.