Recognition: 3 theorem links
· Lean TheoremA Transferable Machine Learning Approach to Predict Optimized Orbitals for Electronic Structure Problems
Pith reviewed 2026-05-08 17:43 UTC · model grok-4.3
The pith
A graph neural network predicts optimized orbital coefficients from molecular geometry, generalizing from small to larger hydrogen systems with small energy errors.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
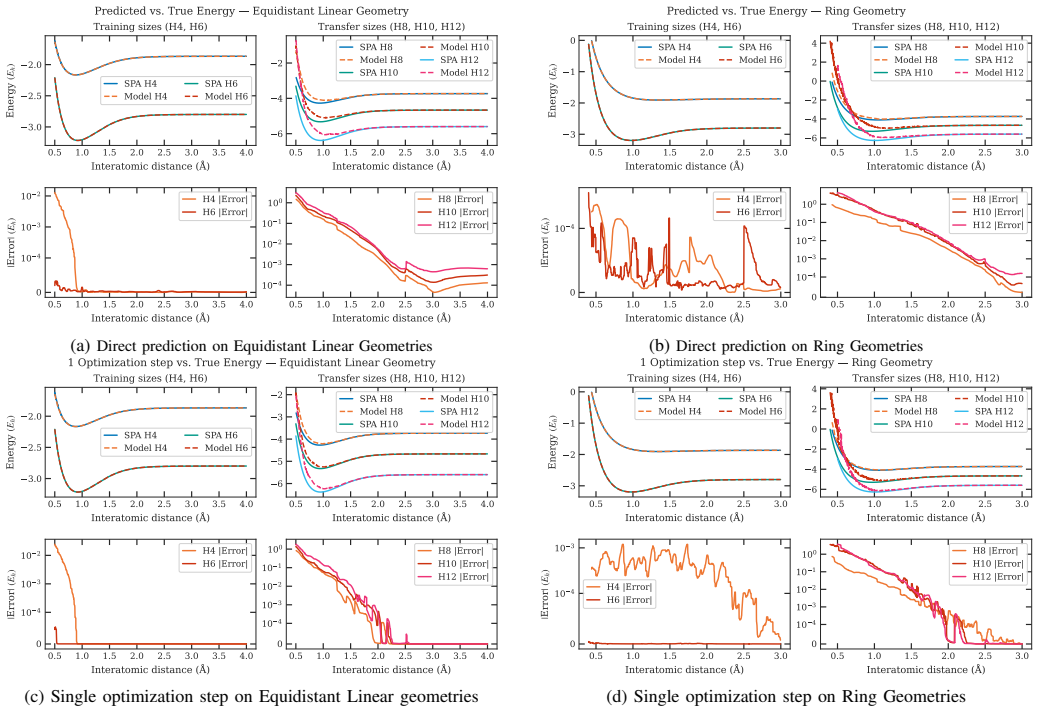
The central discovery is a graph neural network framework that predicts optimized orbital coefficients directly from molecular geometry and pair-wise bonding structure. When trained on tens of thousands of geometries of H4 and H6, the model transfers without retraining to H8, H10, and H12, achieving low energy errors relative to full classical optimization and acting as high-quality initial guesses that accelerate optimizer convergence to the ground state.
What carries the argument
Graph neural network that maps molecular geometry and bonding structure to predicted optimized orbital coefficients for use in variational quantum eigensolver ansatze.
If this is right
- The predicted orbitals allow variational quantum eigensolver calculations on larger systems without performing full classical orbital optimization for each geometry.
- Using the model outputs as initial points substantially reduces the number of optimizer iterations required to reach ground-state energy convergence.
- The transferability to larger systems demonstrates that the approach can scale across chemical space without retraining for each new size.
- Overall, this reduces the classical pre-processing overhead limiting practical deployment of variational quantum eigensolvers on near-term hardware.
Where Pith is reading between the lines
- The same architecture could be tested on molecules containing atoms other than hydrogen to check broader applicability.
- Combining this with other machine learning methods for molecular dynamics might enable real-time orbital predictions during simulations.
- If the generalization holds, it might reduce the need for classical optimization routines entirely in approximate quantum chemistry workflows.
Load-bearing premise
The graph neural network trained on small hydrogenic systems will generate orbital predictions accurate enough for the resulting variational energies and convergence rates to be useful in larger systems.
What would settle it
Measuring the energy error from predicted orbitals on a test set of H12 configurations or a different molecular system and finding deviations significantly exceeding 100 milli-Hartrees would disprove the practical utility claim.
Figures



read the original abstract
Variational quantum eigensolver ans\"atze hold considerable promise for ground-state energy calculations on near-term quantum hardware, yet most promising ansatz designs currently strongly depend on how well the molecular orbital basis captures the electronic correlation of the system. Computing optimized orbital coefficients via classical routines is computationally expensive and must be performed independently for each molecular geometry -- a bottleneck that limits scalability across chemical space. We present a graph neural network framework that predicts optimized orbital coefficients directly from molecular geometry and pair-wise bonding structure. Trained on hydrogenic systems of modest size ($H_4$ and $H_6$) across tens of thousands of geometries, our model transfers to larger, unseen systems ($H_8$, $H_{10}$ and $H_{12}$) without retraining -- demonstrating strong out-of-distribution generalization with respect to system size. When evaluating on structured and random configurations, and comparing against energies obtained with full classical optimization, our model reaches mean absolute energy errors $\mathcal{O}(10^2)$ and $\mathcal{O}(10)$ milli-Hartrees, respectively. Beyond energy estimation, the predicted orbitals serve as high-quality warm-start initializations that substantially reduce optimizer iterations to ground-state energy convergence. These results establish graph neural networks as an effective and scalable strategy for accelerating orbital optimization in hybrid quantum-classical workflows, directly reducing the classical pre-processing overhead that currently limits the practical deployment of variational quantum eigensolver on near-term quantum hardware.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a graph neural network (GNN) trained exclusively on small hydrogen chains (H4 and H6) across tens of thousands of geometries to directly predict optimized orbital coefficients from molecular geometry and bonding information. The model is shown to transfer without retraining to larger unseen chains (H8, H10, H12), yielding mean absolute energy errors of order 100 mHa on structured configurations and 10 mHa on random configurations relative to fully classically optimized orbitals; the predicted orbitals are further claimed to act as effective warm-starts that substantially reduce the number of VQE optimizer iterations needed for convergence.
Significance. If the reported transferability and warm-start benefits hold under detailed scrutiny, the work would address a genuine scalability bottleneck in VQE by replacing per-geometry classical orbital optimization with a single trained model, potentially lowering classical pre-processing costs for near-term quantum chemistry calculations on hydrogenic and related systems.
major comments (2)
- [Abstract] Abstract: the central claim that the GNN produces high-quality warm-start initializations that substantially reduce optimizer iterations lacks quantitative support such as specific iteration counts or convergence curves. Given the O(100) mHa MAE on structured H8-H12 configurations (roughly 1-2% of total energy and exceeding chemical accuracy by >60x), it is unclear if the predictions are close enough to the variational minimum to deliver the claimed speedup.
- [Results] Results section: the manuscript must supply the exact number of test configurations, standard deviations on the reported MAEs, and a breakdown by system size to substantiate the out-of-distribution generalization claim; without these statistics the size-transfer result cannot be assessed for robustness.
minor comments (1)
- [Abstract] Abstract: the encoding of 'ansätze' appears as 'ans'atze'.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed comments, which have helped us identify areas where the manuscript can be strengthened. We provide point-by-point responses below and have revised the manuscript to incorporate the requested clarifications and additional data.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that the GNN produces high-quality warm-start initializations that substantially reduce optimizer iterations lacks quantitative support such as specific iteration counts or convergence curves. Given the O(100) mHa MAE on structured H8-H12 configurations (roughly 1-2% of total energy and exceeding chemical accuracy by >60x), it is unclear if the predictions are close enough to the variational minimum to deliver the claimed speedup.
Authors: We agree that the abstract would benefit from explicit quantitative support for the warm-start claim. While the results section discusses reduced optimizer iterations when using GNN-predicted orbitals, specific counts and curves are not presented there or in the abstract. In the revised manuscript we will update the abstract with representative iteration reductions and add a new figure with convergence curves comparing GNN-initialized VQE runs against standard initializations. This addition will also clarify the practical utility of the observed energy errors: although O(100) mHa exceeds chemical accuracy, the predicted orbitals still capture transferable correlation features that accelerate convergence relative to Hartree-Fock or random starts on these hydrogen chains. revision: yes
-
Referee: [Results] Results section: the manuscript must supply the exact number of test configurations, standard deviations on the reported MAEs, and a breakdown by system size to substantiate the out-of-distribution generalization claim; without these statistics the size-transfer result cannot be assessed for robustness.
Authors: The referee correctly identifies that aggregate MAEs alone are insufficient for assessing robustness. The current manuscript reports overall mean absolute errors for transfer to H8–H12 but omits the exact test-set sizes, standard deviations, and per-system-size breakdowns. We will revise the results section to include these details: the precise number of test configurations per system and configuration type, standard deviations on all reported MAEs, and a table (or bar plot) that breaks down errors separately for H8, H10, and H12 under both structured and random geometries. These additions will allow direct evaluation of the size-transfer generalization. revision: yes
Circularity Check
No circularity: standard supervised ML with held-out OOD evaluation
full rationale
The paper trains a GNN on classically optimized orbitals for H4/H6 geometries and evaluates the model's direct predictions on unseen larger H8/H10/H12 systems by comparing the VQE energies obtained from those predicted orbitals against independent full classical orbital optimization. This constitutes an ordinary train/test split with out-of-distribution testing; the reported MAE values are empirical performance metrics, not quantities forced by construction or by re-expressing the training inputs. No load-bearing self-citations, uniqueness theorems, or ansatze imported from prior author work appear in the provided text, and the central claim (transferable orbital prediction) does not reduce to a tautology or fitted parameter renamed as a prediction.
Axiom & Free-Parameter Ledger
free parameters (1)
- GNN weights and architecture hyperparameters
axioms (1)
- domain assumption Molecular systems can be faithfully represented as graphs with atoms as nodes and bonds as edges for the purpose of orbital prediction
Lean theorems connected to this paper
-
IndisputableMonolith/Cost.lean (J(x) = ½(x+1/x)−1)washburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The primary loss is a Huber loss, computed entry-wise between the predicted generator matrix A_upper and the reference generator A_upper^ref ... L = L_Huber + λ1 L_det + λ2 L_orb.
-
Foundation/AlphaCoordinateFixation.leancost_alpha_one_eq_jcost unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
the orthogonal orbital rotation is recovered via matrix exponentiation, M_oo = e^A.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Simulating physics with computers,
R. P. Feynman, “Simulating physics with computers,” inFeynman and computation. cRc Press, 2018, pp. 133–153
2018
-
[2]
Drug design on quantum computers,
R. Santagati, A. Aspuru-Guzik, R. Babbush, M. Degroote, L. Gonz ´alez, E. Kyoseva, N. Moll, M. Oppel, R. M. Parrish, N. C. Rubinet al., “Drug design on quantum computers,”Nature Physics, vol. 20, no. 4, pp. 549–557, 2024
2024
-
[3]
Quantum simulation of battery materials using ionic pseudopotentials,
M. S. Zini, A. Delgado, R. dos Reis, P. A. M. Casares, J. E. Mueller, A.- C. V oigt, and J. M. Arrazola, “Quantum simulation of battery materials using ionic pseudopotentials,”Quantum, vol. 7, p. 1049, 2023
2023
-
[4]
Simulating key properties of lithium-ion batteries with a fault- tolerant quantum computer,
A. Delgado, P. A. Casares, R. Dos Reis, M. S. Zini, R. Campos, N. Cruz- Hern´andez, A.-C. V oigt, A. Lowe, S. Jahangiri, M. A. Martin-Delgado et al., “Simulating key properties of lithium-ion batteries with a fault- tolerant quantum computer,”Physical Review A, vol. 106, no. 3, p. 032428, 2022
2022
-
[5]
Numerical methods for electronic structure calculations of materials,
Y . Saad, J. R. Chelikowsky, and S. M. Shontz, “Numerical methods for electronic structure calculations of materials,”SIAM review, vol. 52, no. 1, pp. 3–54, 2010
2010
-
[6]
Quantum measurements and the Abelian Stabilizer Problem
A. Y . Kitaev, “Quantum measurements and the abelian stabilizer prob- lem,”arXiv preprint quant-ph/9511026, 1995
work page internal anchor Pith review arXiv 1995
-
[7]
Quantum algorithm providing exponen- tial speed increase for finding eigenvalues and eigenvectors,
D. S. Abrams and S. Lloyd, “Quantum algorithm providing exponen- tial speed increase for finding eigenvalues and eigenvectors,”Physical Review Letters, vol. 83, no. 24, p. 5162, 1999
1999
-
[8]
Simulated quantum computation of molecular energies,
A. Aspuru-Guzik, A. D. Dutoi, P. J. Love, and M. Head-Gordon, “Simulated quantum computation of molecular energies,”Science, vol. 309, no. 5741, pp. 1704–1707, 2005
2005
-
[9]
N. M. Tubman, C. Mejuto-Zaera, J. M. Epstein, D. Hait, D. S. Levine, W. Huggins, Z. Jiang, J. R. McClean, R. Babbush, M. Head-Gordon et al., “Postponing the orthogonality catastrophe: efficient state prepa- ration for electronic structure simulations on quantum devices,”arXiv preprint arXiv:1809.05523, 2018
-
[10]
A variational eigenvalue solver on a photonic quantum processor,
A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik, and J. L. O’brien, “A variational eigenvalue solver on a photonic quantum processor,”Nature communications, vol. 5, no. 1, p. 4213, 2014
2014
-
[11]
Quantum computing in the nisq era and beyond,
J. Preskill, “Quantum computing in the nisq era and beyond,”Quantum, vol. 2, p. 79, 2018
2018
-
[12]
Rayleigh-ritz variational principle for ensembles of fractionally occupied states,
E. K. Gross, L. N. Oliveira, and W. Kohn, “Rayleigh-ritz variational principle for ensembles of fractionally occupied states,”Physical Review A, vol. 37, no. 8, p. 2805, 1988
1988
-
[13]
Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,
J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love, and A. Aspuru-Guzik, “Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz,”Quantum Science and Technology, vol. 4, no. 1, p. 014008, 2019
2019
-
[14]
The variational quantum eigensolver: a review of methods and best practices,
J. Tilly, H. Chen, S. Cao, D. Picozzi, K. Setia, Y . Li, E. Grant, L. Wossnig, I. Rungger, G. H. Boothet al., “The variational quantum eigensolver: a review of methods and best practices,”Physics Reports, vol. 986, pp. 1–128, 2022
2022
-
[15]
A quantum computing view on unitary coupled cluster theory,
A. Anand, P. Schleich, S. Alperin-Lea, P. W. Jensen, S. Sim, M. D ´ıaz- Tinoco, J. S. Kottmann, M. Degroote, A. F. Izmaylov, and A. Aspuru- Guzik, “A quantum computing view on unitary coupled cluster theory,” Chemical Society Reviews, vol. 51, no. 5, pp. 1659–1684, 2022
2022
-
[16]
Optimized low-depth quantum circuits for molecular electronic structure using a separable-pair approx- imation,
J. S. Kottmann and A. Aspuru-Guzik, “Optimized low-depth quantum circuits for molecular electronic structure using a separable-pair approx- imation,”Physical Review A, vol. 105, no. 3, p. 032449, 2022
2022
-
[17]
Orbital optimized unitary coupled cluster theory for quantum computer,
W. Mizukami, K. Mitarai, Y . O. Nakagawa, T. Yamamoto, T. Yan, and Y .-y. Ohnishi, “Orbital optimized unitary coupled cluster theory for quantum computer,”Physical Review Research, vol. 2, no. 3, p. 033421, 2020
2020
-
[18]
Quantum orbital-optimized unitary cou- pled cluster methods in the strongly correlated regime: Can quantum algorithms outperform their classical equivalents?
I. O. Sokolov, P. K. Barkoutsos, P. J. Ollitrault, D. Greenberg, J. Rice, M. Pistoia, and I. Tavernelli, “Quantum orbital-optimized unitary cou- pled cluster methods in the strongly correlated regime: Can quantum algorithms outperform their classical equivalents?”The Journal of chemical physics, vol. 152, no. 12, 2020
2020
-
[19]
A state-averaged orbital-optimized hybrid quantum–classical al- gorithm for a democratic description of ground and excited states,
S. Yalouz, B. Senjean, J. G ¨unther, F. Buda, T. E. O’Brien, and L. Viss- cher, “A state-averaged orbital-optimized hybrid quantum–classical al- gorithm for a democratic description of ground and excited states,” Quantum Science and Technology, vol. 6, no. 2, p. 024004, 2021
2021
-
[20]
Graph neural networks for materials science and chemistry,
P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. van Hoesel, H. Schopmans, T. Sommeret al., “Graph neural networks for materials science and chemistry,”Communications Materials, vol. 3, no. 1, p. 93, 2022
2022
-
[21]
Convolutional networks on graphs for learning molecular fingerprints,
D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik, and R. P. Adams, “Convolutional networks on graphs for learning molecular fingerprints,”Advances in neural information processing systems, vol. 28, 2015
2015
-
[22]
Neural message passing for quantum chemistry,
J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals, and G. E. Dahl, “Neural message passing for quantum chemistry,” inInternational conference on machine learning. Pmlr, 2017, pp. 1263–1272
2017
-
[23]
Schnet: A continuous-filter convo- lutional neural network for modeling quantum interactions,
K. Sch ¨utt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko, and K.-R. M ¨uller, “Schnet: A continuous-filter convo- lutional neural network for modeling quantum interactions,”Advances in neural information processing systems, vol. 30, 2017
2017
-
[24]
Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges,
O. T. Unke and M. Meuwly, “Physnet: A neural network for predicting energies, forces, dipole moments, and partial charges,”Journal of chemical theory and computation, vol. 15, no. 6, pp. 3678–3693, 2019
2019
-
[25]
Directional message passing for molecular graphs,
J. Gasteiger, J. Groß, and S. G ¨unnemann, “Directional message passing for molecular graphs,” inInternational Conference on Learning Repre- sentations, 2020
2020
-
[26]
Gemnet: Universal di- rectional graph neural networks for molecules,
J. Gasteiger, F. Becker, and S. G ¨unnemann, “Gemnet: Universal di- rectional graph neural networks for molecules,”Advances in Neural Information Processing Systems, vol. 34, pp. 6790–6802, 2021
2021
-
[27]
The reduction of a graph to canonical form and the algebra which appears therein,
B. Weisfeiler and A. Leman, “The reduction of a graph to canonical form and the algebra which appears therein,”nti, Series, vol. 2, no. 9, pp. 12–16, 1968
1968
-
[28]
Molecule graph networks with many-body equivariant interactions,
Z. Mao, C.-S. Hu, J. Li, C. Liang, D. Das, M. Sumita, K. Xia, and K. Tsuda, “Molecule graph networks with many-body equivariant interactions,”Journal of Chemical Theory and Computation, vol. 21, no. 16, pp. 7954–7966, 2025
2025
-
[29]
Recipe for a general, powerful, scalable graph transformer,
L. Ramp ´aˇsek, M. Galkin, V . P. Dwivedi, A. T. Luu, G. Wolf, and D. Beaini, “Recipe for a general, powerful, scalable graph transformer,” Advances in Neural Information Processing Systems, vol. 35, pp. 14 501–14 515, 2022
2022
-
[30]
Learning interactions between rydberg atoms,
O. Simard, A. Dawid, J. Tindall, M. Ferrero, A. M. Sengupta, and A. Georges, “Learning interactions between rydberg atoms,”PRX Quan- tum, vol. 6, no. 3, p. 030324, 2025
2025
-
[31]
D. Bincoletto, K. Stein, J. Motyl, and J. S. Kottmann, “A transferable machine learning approach to predict quantum circuit parameters for electronic structure problems,”arXiv preprint arXiv:2511.03726, 2025
-
[32]
quanti-gin: A customizable data generator for quantum simulation,
K. Stein and T. Truong, “quanti-gin: A customizable data generator for quantum simulation,” 2025, gitHub repository. [Online]. Available: https://github.com/nylser/quanti-gin
2025
-
[33]
Tequila: A platform for rapid development of quantum algorithms,
J. S. Kottmann, S. Alperin-Lea, T. Tamayo-Mendoza, A. Cervera- Lierta, C. Lavigne, T.-C. Yen, V . Verteletskyi, P. Schleich, A. Anand, M. Degrooteet al., “Tequila: A platform for rapid development of quantum algorithms,”Quantum Science and Technology, vol. 6, no. 2, p. 024009, 2021
2021
-
[34]
Molecular quantum circuit design: A graph-based approach,
J. S. Kottmann, “Molecular quantum circuit design: A graph-based approach,”Quantum, vol. 7, p. 1073, 2023
2023
-
[35]
B. S. Kang, V . C. Bhethanabotla, A. Tavakoli, M. D. Hanisch, W. A. Goddard III, and A. Anandkumar, “Orbitall: a unified quantum mechan- ical representation deep learning framework for all molecular systems,” arXiv preprint arXiv:2507.03853, 2025
-
[36]
Casnet: Learning complete active space orbitals using message passing neural networks,
R. Van Workum, J. Malhado, and P. Marquetand, “Casnet: Learning complete active space orbitals using message passing neural networks,” chemrxiv-2023-lwj87, 2023
2023
-
[37]
V . P. Dwivedi, A. T. Luu, T. Laurent, Y . Bengio, and X. Bresson, “Graph neural networks with learnable structural and positional representations,” arXiv preprint arXiv:2110.07875, 2021
-
[38]
Dynamic edge-conditioned filters in convolutional neural networks on graphs,
M. Simonovsky and N. Komodakis, “Dynamic edge-conditioned filters in convolutional neural networks on graphs,” inProceedings of the IEEE conference on computer vision and pattern recognition, 2017, pp. 3693– 3702
2017
-
[39]
Robust estimation of a location parameter,
P. J. Huber, “Robust estimation of a location parameter,” inBreak- throughs in statistics: Methodology and distribution. Springer, 1992, pp. 492–518
1992
-
[40]
Direct determination of optimal pair-natural orbitals in a real-space representation: The second- order Moller–Plesset energy,
J. S. Kottmann, F. A. Bischoff, and E. F. Valeev, “Direct determination of optimal pair-natural orbitals in a real-space representation: The second- order Moller–Plesset energy,”The Journal of Chemical Physics, vol. 152, no. 7, p. 074105, 2020, publisher: AIP Publishing LLC
2020
-
[41]
Direct determination of optimal real-space orbitals for correlated electronic structure of molecules,
E. F. Valeev, R. J. Harrison, A. A. Holmes, C. C. Peterson, and D. A. Penchoff, “Direct determination of optimal real-space orbitals for correlated electronic structure of molecules,”Journal of Chemical Theory and Computation, vol. 19, no. 20, pp. 7230–7241, 2023
2023
-
[42]
The advent of fully variational quantum eigensolvers using a hybrid multiresolution approach,
F. Langkabel, S. Knecht, and J. S. Kottmann, “The advent of fully variational quantum eigensolvers using a hybrid multiresolution approach,” 2025. [Online]. Available: https://arxiv.org/abs/2410.19116
-
[43]
Quantum algorithmic approach to multi- configurational valence bond theory: Insights from interpretable circuit design,
J. S. Kottmann and F. Scala, “Quantum algorithmic approach to multi- configurational valence bond theory: Insights from interpretable circuit design,”Journal of Chemical Theory and Computation, vol. 20, no. 9, pp. 3514–3523, 2024
2024
-
[44]
Seniority-based coupled cluster theory,
T. M. Henderson, I. W. Bulik, T. Stein, and G. E. Scuseria, “Seniority-based coupled cluster theory,”The Journal of Chemical Physics, vol. 141, no. 24, p. 244104, Dec 2014. [Online]. Available: http://dx.doi.org/10.1063/1.4904384
-
[45]
Mapping Molecular Orbital—Configuration Interaction to Valence Bond Wave Functions,
S. Shaik and P. C. Hiberty, “Mapping Molecular Orbital—Configuration Interaction to Valence Bond Wave Functions,” inA Chemist’s Guide to Valence Bond Theory. John Wiley & Sons, Ltd, 2007, ch. 4, pp. 81–93
2007
-
[46]
Accurate and gate-efficient quantum Ans\
H. G. A. Burton, “Accurate and gate-efficient quantum Ans\”atze for electronic states without adaptive optimization,”Physical Review Research, vol. 6, no. 2, p. 023300, Jun. 2024
2024
-
[47]
Lo- cal, expressive, quantum-number-preserving VQE ans ¨atze for fermionic systems,
G.-L. R. Anselmetti, D. Wierichs, C. Gogolin, and R. M. Parrish, “Lo- cal, expressive, quantum-number-preserving VQE ans ¨atze for fermionic systems,”New Journal of Physics, vol. 23, no. 11, p. 113010, Nov. 2021. APPENDIX APPENDIXA SEPARABLEPAIRAPPROXIMATION The Separable Pair Approximation (SPA) [16] is a varia- tional circuit design that has demonstrate...
2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.