Recognition: unknown
Density diversity in training data governs thermodynamic transferability of machine learning interatomic potentials
Pith reviewed 2026-05-08 04:16 UTC · model grok-4.3
The pith
Diversifying density in training data produces machine learning interatomic potentials that transfer across thermodynamic states.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
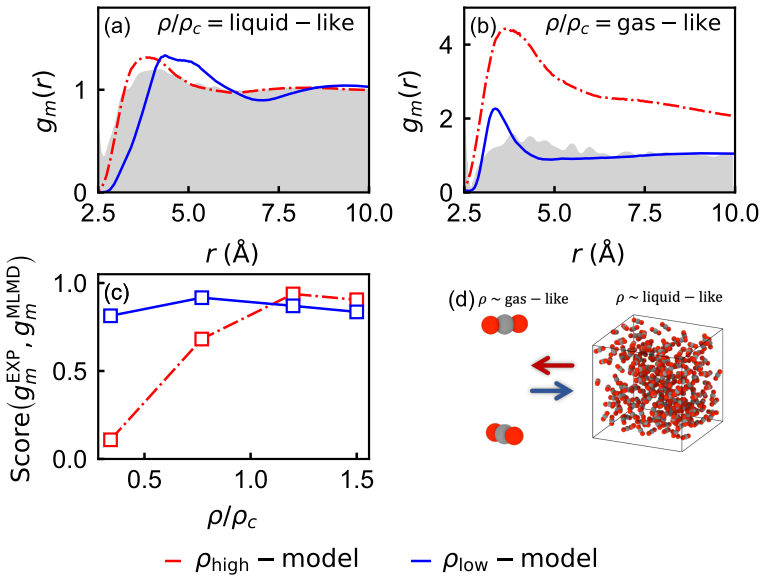
Diversifying the density of training configurations, rather than temperature, is the most effective strategy for building thermodynamically transferable MLIPs within a fixed computational budget. Foundation MLIPs trained on solid-state databases accurately describe liquid-like densities but fail at gas-like conditions, while molecular-database-trained models exhibit the opposite behavior. Controlled from-scratch training and distillation experiments confirm that density-diverse datasets resolve both failure modes, whereas temperature-diverse datasets cannot compensate for missing density regimes. Coordination number analysis reveals the physical origin of this behavior: local coordination is
What carries the argument
Density diversity in the training dataset, which increases the range of local atomic coordination environments more effectively than temperature diversity.
Where Pith is reading between the lines
- The same density-first sampling principle may improve transferability in ML potentials trained for other state variables that strongly affect local structure, such as composition.
- Models for inhomogeneous systems like interfaces could be built by deliberately sampling across density gradients rather than uniform temperature sweeps.
- The validation framework based on density coverage offers a practical checklist for assessing existing foundation models before deployment in variable-pressure fluid simulations.
Load-bearing premise
The coordination number analysis and observed failure modes in the tested foundation and from-scratch models apply to other MLIP architectures and chemical systems.
What would settle it
Repeat the from-scratch training and distillation experiments on a different fluid system such as water and test whether density diversity still outperforms temperature diversity when evaluating transfer errors at densities outside the training range.
Figures





read the original abstract
Machine learning interatomic potentials (MLIPs) offer first-principles accuracy with reduced computational cost, but their transferability across different thermodynamic states remains questionable, particularly for fluid systems where molecules experience local environments far from crystalline equilibrium. Here, we demonstrate that diversifying the density of training configurations, rather than temperature, is the most effective strategy for building thermodynamically transferable MLIPs within a fixed computational budget. We first show that foundation MLIPs trained on solid-state databases accurately describe liquid-like densities but fail at gas-like conditions, while molecular-database-trained models exhibit the opposite behavior. Controlled from-scratch training and distillation experiments confirm that density-diverse datasets resolve both failure modes, whereas temperature-diverse datasets cannot compensate for missing density regimes. Coordination number analysis reveals the physical origin of this behavior: local coordination topology is more susceptible to density than temperature, leading to further structural diversity. These results establish density diversity as a design principle for thermodynamically transferable MLIPs and provide a validation framework for assessing the thermodynamic coverage of both foundation and from-scratch models, enabling reliable atomistic simulation of fluid-phase processes across diverse operating conditions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper claims that for machine learning interatomic potentials (MLIPs) applied to fluid systems, diversifying the density of training configurations is more effective than diversifying temperature for achieving thermodynamic transferability within a fixed computational budget. This is supported by showing that foundation models trained on solid-state databases succeed at liquid-like densities but fail at gas-like conditions (and vice versa for molecular-database models), with controlled from-scratch training and distillation experiments demonstrating that density-diverse datasets resolve both failure modes while temperature-diverse ones do not. Coordination-number analysis is presented as the physical explanation, since local coordination topology varies more strongly with density than with temperature.
Significance. If the central result holds, the work supplies a concrete, testable design rule for constructing thermodynamically transferable MLIPs and a validation framework based on density coverage. This is practically useful for fluid-phase simulations in chemistry and materials science, where operating conditions often span wide density ranges. The controlled experiments and coordination-number interpretation add interpretability and falsifiability that many MLIP transferability studies lack.
major comments (3)
- [Results and Discussion sections] The generalization claim that density diversity is the governing factor (abstract and final paragraph) rests on experiments performed only for the specific foundation models (solid-state vs. molecular databases) and from-scratch/distillation setups described. No additional architectures (different message-passing depths, cutoff schemes, or equivariant layers) or chemically dissimilar systems are tested, so it remains unclear whether coordination number remains the dominant transferable descriptor outside the examined cases.
- [Experimental results on foundation models and controlled trainings] Quantitative support for the failure modes and their resolution is insufficiently detailed. The abstract and results describe qualitative success/failure but do not report dataset sizes, model architectures, error bars, or specific error metrics (e.g., energy/force RMSE or structural deviation thresholds) that would allow assessment of effect size and statistical significance.
- [Coordination number analysis subsection] The coordination-number analysis is offered as the physical origin, yet the manuscript does not compare it against other structural descriptors (e.g., radial distribution functions, angular distributions, or ring statistics) to establish that density-induced changes in coordination are the primary driver rather than one of several correlated factors.
minor comments (2)
- [Figures] Figure captions and axis labels should explicitly state the number of configurations, temperature/density ranges, and error metrics used in each panel to improve reproducibility.
- [Abstract and Conclusions] The term 'parameter-free' or similar phrasing for the design principle should be avoided or qualified, since the conclusion is drawn from empirical comparisons rather than a derivation independent of the chosen models and systems.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback and for highlighting the practical value of our results. Below we respond point by point to the major comments, providing clarifications from the manuscript and indicating the revisions we will implement.
read point-by-point responses
-
Referee: [Results and Discussion sections] The generalization claim that density diversity is the governing factor (abstract and final paragraph) rests on experiments performed only for the specific foundation models (solid-state vs. molecular databases) and from-scratch/distillation setups described. No additional architectures (different message-passing depths, cutoff schemes, or equivariant layers) or chemically dissimilar systems are tested, so it remains unclear whether coordination number remains the dominant transferable descriptor outside the examined cases.
Authors: We agree that the experiments are confined to the models and chemical systems described and do not demonstrate universality. The controlled from-scratch and distillation protocols were deliberately chosen to isolate density versus temperature effects while holding other variables fixed. The coordination-number analysis supplies a physically interpretable mechanism, but we do not claim it is the sole descriptor in all cases. In the revised manuscript we will moderate the language in the abstract and concluding paragraph, add an explicit Limitations subsection that states the current scope, and outline the need for validation on additional architectures and dissimilar chemistries. revision: yes
-
Referee: [Experimental results on foundation models and controlled trainings] Quantitative support for the failure modes and their resolution is insufficiently detailed. The abstract and results describe qualitative success/failure but do not report dataset sizes, model architectures, error bars, or specific error metrics (e.g., energy/force RMSE or structural deviation thresholds) that would allow assessment of effect size and statistical significance.
Authors: Dataset sizes, model architectures, training protocols, and error metrics (including energy/force RMSE with standard deviations from repeated runs) are reported in the Methods section and Supplementary Information. To improve accessibility we will extract the key quantitative values and statistical details into the main Results text, add error bars to the relevant figures, and include explicit numerical thresholds for the success/failure criteria used in the transferability tests. revision: yes
-
Referee: [Coordination number analysis subsection] The coordination-number analysis is offered as the physical origin, yet the manuscript does not compare it against other structural descriptors (e.g., radial distribution functions, angular distributions, or ring statistics) to establish that density-induced changes in coordination are the primary driver rather than one of several correlated factors.
Authors: Coordination number was highlighted because it exhibits the largest variation across the density range examined and correlates directly with the observed transferability failures. We recognize that other descriptors are correlated with density. In the revised manuscript we will add a short comparative analysis that includes radial distribution functions and angular distributions, demonstrating that coordination number provides the strongest discrimination between the density regimes where transferability succeeds or fails. revision: yes
Circularity Check
No circularity; results rest on comparative experiments and coordination analysis
full rationale
The paper advances its central claim through controlled from-scratch training, distillation, and foundation-model evaluation experiments that directly compare density-diverse versus temperature-diverse datasets, with coordination-number statistics offered as the physical explanation for observed transferability differences. No equations, fitted parameters renamed as predictions, or self-citation chains are invoked to derive the result; the findings are presented as empirical outcomes that can be replicated or falsified on the described systems and architectures. The derivation chain is therefore self-contained and does not reduce to its inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Local atomic coordination topology is the dominant physical feature controlling thermodynamic transferability of interatomic potentials.
Reference graph
Works this paper leans on
-
[1]
Simeski, M
F. Simeski, M. Ihme, Supercritical fluids behave as complex networks, Nature Communications 14 (1) (2023) 1996
2023
-
[2]
T. J. Yoon, M. Y. Ha, W. B. Lee, Y.-W. Lee, Molecular dynamics sim- ulation on the local density distribution and solvation structure of su- percritical co2 around naphthalene, The Journal of Supercritical Fluids 130 (2017) 364–372
2017
-
[3]
J. Li, Y. Lang, B. Li, H. Zhang, J. Zhang, S. S. Rahman, Molecular mechanism of supercritical co2 enhancing shale oil production by ex- traction characteristics, Langmuir 41 (17) (2025) 11147–11160
2025
-
[4]
Y. Han, B. Zhang, J. Jiang, L. Cen, J. Li, L. Zhao, Z. Xi, Understand- ing the enhancement of entrainers on the supercritical carbon dioxide extraction of phospholipids: A molecular dynamics simulation, Journal of Molecular Liquids 411 (2024) 125647
2024
-
[5]
Muralidharan, M
A. Muralidharan, M. I. Chaudhari, L. R. Pratt, S. B. Rempe, Molecular dynamics of lithium ion transport in a model solid electrolyte interphase, Scientific reports 8 (1) (2018) 10736
2018
-
[6]
C. Park, S. Kim, S. Kim, M. Lee, S. Kim, J. Cho, A. Park, S. Kwon, M. Kim, S. Rho, et al., Lithium solvation structure and dynamics in an ionic liquid electrolyte: A deep learning-assisted analysis on polar- izable molecular dynamics simulations, Chemical Engineering Journal 508 (2025) 160662. 31
2025
-
[7]
Kubisiak, A
P. Kubisiak, A. Eilmes, Molecular dynamics simulations of ionic liquid based electrolytes for na-ion batteries: Effects of force field, The Journal of Physical Chemistry B 121 (42) (2017) 9957–9968
2017
-
[8]
J. J. Winetrout, K. Kanhaiya, J. Kemppainen, P. J. in ‘t Veld, G. Sachdeva, R. Pandey, B. Damirchi, A. van Duin, G. M. Odegard, H. Heinz, Implementing reactivity in molecular dynamics simulations with harmonic force fields, Nature communications 15 (1) (2024) 7945
2024
-
[9]
Q. Mao, M. Feng, X. Z. Jiang, Y. Ren, K. H. Luo, A. C. van Duin, Classical and reactive molecular dynamics: Principles and applications in combustion and energy systems, Progress in Energy and Combustion Science 97 (2023) 101084
2023
-
[10]
J. Choi, B. Jun, Y. Jung, Reliable li-ion conductivity with efficient data generation and uncertainty estimation toward large-scale screen- ing, Chemical Engineering Journal 516 (2025) 163847
2025
-
[11]
Zhang, Z
P. Zhang, Z. Liu, X. Wang, R. Mi, Y. Pang, F. Pan, J. Li, Z. Wang, W. Wang, K. H. Luo, Unraveling the early-stage hydration mechanism of tricalcium silicate via machine learning potentials, Chemical Engi- neering Journal (2026) 176856
2026
-
[12]
O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K.T.Schutt, A.Tkatchenko, K.-R.Muller, Machinelearningforcefields, Chemical reviews 121 (16) (2021) 10142–10186
2021
-
[13]
M. Yang, L. Bonati, D. Polino, M. Parrinello, Using metadynamics to 32 build neural network potentials for reactive events: the case of urea decomposition in water, Catalysis Today 387 (2022) 143–149
2022
-
[14]
N. V. Avula, M. L. Klein, S. Balasubramanian, Understanding the anomalous diffusion of water in aqueous electrolytes using machine learned potentials, The Journal of Physical Chemistry Letters 14 (42) (2023) 9500–9507
2023
-
[15]
M. Kim, S. Kim, C. Hyeon, J. W. Yu, S. Q. Choi, W. B. Lee, Anomalous water penetration in al3+ dissolution, The Journal of Physical Chem- istry Letters 15 (43) (2024) 10903–10908
2024
-
[16]
J. H. Ryu, J. W. Yu, T. J. Yoon, W. B. Lee, Understanding the dielectric relaxation of liquid water using neural network potential and classical pairwise potential, Journal of Molecular Liquids 397 (2024) 124054
2024
-
[17]
J. H. Ryu, S. Kim, M. Kim, J. W. Yu, T. J. Yoon, W. B. Lee, Explor- ing the reaction network of acetic acid in supercritical water via ma- chine learning interatomic potential, Journal of Chemical Information and Modeling 65 (16) (2025) 8614–8623
2025
-
[18]
J. W. Yu, S. Kim, J. H. Ryu, W. B. Lee, T. J. Yoon, Spatiotemporal characterization of water diffusion anomalies in saline solutions using machine learning force field, Science Advances 10 (50) (2024) eadp9662
2024
-
[19]
J. Zeng, D. Zhang, A. Peng, X. Zhang, S. He, Y. Wang, X. Liu, H. Bi, Y. Li, C. Cai, et al., Deepmd-kit v3: a multiple-backend framework for machine learning potentials, Journal of chemical theory and computa- tion 21 (9) (2025) 4375–4385. 33
2025
-
[20]
Molinari, T
S.Batzner, A.Musaelian, L.Sun, M.Geiger, J.P.Mailoa, M.Kornbluth, N. Molinari, T. E. Smidt, B. Kozinsky, E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials, Nature communications 13 (1) (2022) 2453
2022
-
[21]
Batatia, D
I. Batatia, D. P. Kovacs, G. N. C. Simm, C. Ortner, G. Csanyi, MACE: Higher order equivariant message passing neural networks for fast and accurate force fields, in: A. H. Oh, A. Agarwal, D. Belgrave, K. Cho (Eds.), Advances in Neural Information Processing Systems, 2022. URLhttps://openreview.net/forum?id=YPpSngE-ZU
2022
-
[22]
Y. Park, J. Kim, S. Hwang, S. Han, Scalable parallel algorithm for graph neural network interatomic potentials in molecular dynamics sim- ulations, J. Chem. Theory Comput. 20 (11) (2024) 4857–4868.doi: 10.1021/acs.jctc.4c00190
-
[23]
B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel, G. Ceder, Chgnet as a pretrained universal neural network potential for charge- informed atomistic modelling, Nature Machine Intelligence 5 (9) (2023) 1031–1041
2023
-
[24]
I.Batatia, P.Benner, Y.Chiang, A.M.Elena, D.P.Kovács, J.Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, et al., A founda- tion model for atomistic materials chemistry, The Journal of chemical physics 163 (18) (2025)
2025
-
[25]
J. Kim, J. Kim, J. Kim, J. Lee, Y. Park, Y. Kang, S. Han, Data- efficient multifidelity training for high-fidelity machine learning inter- 34 atomic potentials, J. Am. Chem. Soc. 147 (1) (2024) 1042–1054.doi: 10.1021/jacs.4c14455
-
[26]
Kabylda, J
A. Kabylda, J. T. Frank, S. Suárez-Dou, A. Khabibrakhmanov, L. Medrano Sandonas, O. T. Unke, S. Chmiela, K.-R. Muüller, A. Tkatchenko, Molecular simulations with a pretrained neural network and universal pairwise force fields, Journal of the American Chemical Society 147 (37) (2025) 33723–33734
2025
-
[27]
Bowen, Materials Project Trajectory (MPtrj) Dataset
B. Deng, Materials Project Trajectory (MPtrj) Dataset (7 2023). doi:10.6084/m9.figshare.23713842.v2. URLhttps://figshare.com/articles/dataset/Materials_ Project_Trjectory_MPtrj_Dataset/23713842
-
[28]
M. M. Ghahremanpour, P. J. Van Maaren, D. Van Der Spoel, The alexandria library, a quantum-chemical database of molecular properties for force field development, Scientific data 5 (1) (2018) 1–10
2018
-
[29]
Barroso-Luque et al., Open Materials 2024 (OMat24) Inorganic Materials Dataset and Models
L. Barroso-Luque, M. Shuaibi, X. Fu, B. M. Wood, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick, Z. W. Ulissi, Open materials 2024 (omat24) in- organic materials dataset and models, arXiv preprint arXiv:2410.12771 (2024)
-
[30]
M. Y. Ha, T. J. Yoon, T. Tlusty, Y. Jho, W. B. Lee, Widom delta of supercritical gas–liquid coexistence, The journal of physical chemistry letters 9 (7) (2018) 1734–1738
2018
- [31]
-
[32]
Grunert, M
M. Grunert, M. Großmann, J. Hänseroth, A. Flötotto, J. Oumard, J. L. Wolf, E. Runge, C. Dreßler, Modeling complex proton trans- port phenomena—exploring the limits of fine-tuning and transferability of foundational machine-learned force fields, The Journal of Physical Chemistry C 129 (21) (2025) 9662–9669
2025
-
[33]
S. Ju, J. You, G. Kim, Y. Park, H. An, S. Han, Application of pre- trained universal machine-learning interatomic potential for physico- chemical simulation of liquid electrolytes in li-ion batteries, Digital Dis- covery 4 (6) (2025) 1544–1559
2025
-
[34]
Z. A. Goodwin, M. B. Wenny, J. H. Yang, A. Cepellotti, J. Ding, K. Bystrom, B. R. Duschatko, A. Johansson, L. Sun, S. Batzner, et al., Transferability and accuracy of ionic liquid simulations with equivari- ant machine learning interatomic potentials, The Journal of Physical Chemistry Letters 15 (30) (2024) 7539–7547
2024
-
[35]
W. G. Stark, C. van der Oord, I. Batatia, Y. Zhang, B. Jiang, G. Csányi, R. J. Maurer, Benchmarking of machine learning interatomic potentials for reactive hydrogen dynamics at metal surfaces, Machine Learning: Science and Technology 5 (3) (2024) 030501
2024
-
[36]
O. T. Unke, M. Stöhr, S. Ganscha, T. Unterthiner, H. Maennel, S.Kashubin, D.Ahlin, M.Gastegger, L.MedranoSandonas, J.T.Berry- man, et al., Biomolecular dynamics with machine-learned quantum- 36 mechanical force fields trained on diverse chemical fragments, Science Advances 10 (14) (2024) eadn4397
2024
-
[37]
J. Hoja, L. Medrano Sandonas, B. G. Ernst, A. Vazquez-Mayagoitia, R. A. DiStasio Jr, A. Tkatchenko, Qm7-x, a comprehensive dataset of quantum-mechanical properties spanning the chemical space of small organic molecules, Scientific data 8 (1) (2021) 43
2021
-
[38]
Medrano Sandonas, D
L. Medrano Sandonas, D. Van Rompaey, A. Fallani, M. Hilfiker, D. Hahn, L. Perez-Benito, J. Verhoeven, G. Tresadern, J. Kurt Weg- ner, H. Ceulemans, et al., Dataset for quantum-mechanical exploration of conformers and solvent effects in large drug-like molecules, Scientific Data 11 (1) (2024) 742
2024
-
[39]
Eastman, P
P. Eastman, P. K. Behara, D. L. Dotson, R. Galvelis, J. E. Herr, J. T. Horton, Y.Mao, J.D.Chodera, B.P.Pritchard, Y.Wang, etal., Spice, a dataset of drug-like molecules and peptides for training machine learning potentials, Scientific Data 10 (1) (2023) 11
2023
-
[40]
A. G. Donchev, A. G. Taube, E. Decolvenaere, C. Hargus, R. T. McGib- bon, K.-H. Law, B. A. Gregersen, J.-L. Li, K. Palmo, K. Siva, et al., Quantum chemical benchmark databases of gold-standard dimer inter- action energies, Scientific data 8 (1) (2021) 55
2021
-
[41]
J. P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approxima- tion made simple, Physical review letters 77 (18) (1996) 3865
1996
-
[42]
V. I. Anisimov, J. Zaanen, O. K. Andersen, Band theory and mott 37 insulators: Hubbard u instead of stoner i, Physical Review B 44 (3) (1991) 943
1991
-
[43]
Adamo, M
C. Adamo, M. Cossi, V. Barone, An accurate density functional method for the study of magnetic properties: the pbe0 model, Journal of Molec- ular Structure: THEOCHEM 493 (1-3) (1999) 145–157
1999
-
[44]
Tkatchenko, R
A. Tkatchenko, R. A. DiStasio Jr, R. Car, M. Scheffler, Accurate and efficient method for many-body van der waals interactions, Physical re- view letters 108 (23) (2012) 236402
2012
-
[45]
Frank, O
T. Frank, O. Unke, K.-R. Müller, So3krates: Equivariant attention for interactions on arbitrary length-scales in molecular systems, Advances in Neural Information Processing Systems 35 (2022) 29400–29413
2022
-
[46]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (dft- d) for the 94 elements h-pu, The Journal of chemical physics 132 (15) (2010)
2010
-
[47]
Grimme, S
S. Grimme, S. Ehrlich, L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, Journal of computational chemistry 32 (7) (2011) 1456–1465
2011
-
[48]
A. Dunn, Q. Wang, A. Ganose, D. Dopp, A. Jain, Benchmarking mate- rials property prediction methods: the matbench test set and automat- miner reference algorithm, npj Computational Materials 6 (1) (2020) 138. 38
2020
-
[49]
Cipriani, M
P. Cipriani, M. Nardone, F. Ricci, Neutron diffraction measurements on co2 in both undercritical and supercritical states, Physica B: Condensed Matter 241 (1997) 940–946
1997
-
[50]
J. G. Harris, K. H. Yung, Carbon dioxide’s liquid-vapor coexistence curve and critical properties as predicted by a simple molecular model, The Journal of Physical Chemistry 99 (31) (1995) 12021–12024
1995
-
[51]
L. B. Skinner, C. Huang, D. Schlesinger, L. G. Pettersson, A. Nilsson, C. J. Benmore, Benchmark oxygen-oxygen pair-distribution function of ambient water from x-ray diffraction measurements with a wide q-range, The Journal of chemical physics 138 (7) (2013)
2013
-
[52]
Riera, E
M. Riera, E. P. Yeh, F. Paesani, Data-driven many-body models for molecular fluids: Co2/h2o mixtures as a case study, Journal of Chemical Theory and Computation 16 (4) (2020) 2246–2257
2020
-
[53]
S.Yue, M.Riera, R.Ghosh, A.Z.Panagiotopoulos, F.Paesani, Transfer- ability of data-driven, many-body models for co2 simulations in the va- por and liquid phases, The Journal of Chemical Physics 156 (10) (2022)
2022
-
[54]
J. Kim, J. You, Y. Park, Y. Lim, Y. Kang, J. Kim, H. Jeon, S. Ju, D. Hong, S. Y. Lee, S. Choi, Y. Kim, J. W. Lee, S. Han, Optimizing cross-domain transfer for universal machine learning interatomic poten- tials, arXiv (2025).doi:10.48550/arxiv.2510.11241
-
[55]
T. J. Yoon, M. Y. Ha, E. A. Lazar, W. B. Lee, Y.-W. Lee, Topological characterization of rigid–nonrigid transition across the frenkel line, The Journal of Physical Chemistry Letters 9 (22) (2018) 6524–6528. 39
2018
-
[56]
T. J. Yoon, M. Y. Ha, W. B. Lee, Y.-W. Lee, E. A. Lazar, Topological generalization of the rigid-nonrigid transition in soft-sphere and hard- sphere fluids, Physical Review E 99 (5) (2019) 052603
2019
-
[57]
T. J. Yoon, M. Y. Ha, E. A. Lazar, W. B. Lee, Y.-W. Lee, Topological extension of the isomorph theory based on the shannon entropy, Physical Review E 100 (1) (2019) 012118
2019
-
[58]
Behler, M
J. Behler, M. Parrinello, Generalized neural-network representation of high-dimensional potential-energy surfaces, Physical review letters 98 (14) (2007) 146401
2007
-
[59]
Hjorth Larsen, J
A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Har- gus, et al., The atomic simulation environment—a python library for working with atoms, Journal of Physics: Condensed Matter 29 (27) (2017) 273002
2017
-
[60]
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakat- suji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goin...
2016
-
[61]
Weigend, R
F. Weigend, R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for h to rn: Design and assessment of accuracy, Physical Chemistry Chemical Physics 7 (18) (2005) 3297–3305
2005
-
[62]
G. J. Martyna, A. Hughes, M. E. Tuckerman, Molecular dynamics algo- rithms for path integrals at constant pressure, The Journal of chemical physics 110 (7) (1999) 3275–3290
1999
-
[63]
M. E. Tuckerman, Statistical mechanics: theory and molecular simula- tion, Oxford university press, 2023
2023
-
[64]
S. Takamoto, C. Shinagawa, D. Motoki, K. Nakago, W. Li, I. Kurata, T. Watanabe, Y. Yayama, H. Iriguchi, Y. Asano, T. Onodera, T. Ishii, T. Kudo, H. Ono, R. Sawada, R. Ishitani, M. Ong, T. Yamaguchi, T. Kataoka, A. Hayashi, T. Ibuka, Pfp: Universal neural network po- tential for material discovery (2021).arXiv:2106.14583
-
[65]
Martínez, R
L. Martínez, R. Andrade, E. G. Birgin, J. M. Martínez, Packmol: A package for building initial configurations for molecular dynamics simu- lations, Journal of computational chemistry 30 (13) (2009) 2157–2164. 41
2009
-
[66]
Riera, C
M. Riera, C. Knight, E. F. Bull-Vulpe, X. Zhu, H. Agnew, D. G. Smith, A. C. Simmonett, F. Paesani, Mbx: A many-body energy and force cal- culatorfordata-drivenmany-bodysimulations, TheJournalofChemical Physics 159 (5) (2023)
2023
-
[67]
Ceriotti, J
M. Ceriotti, J. More, D. E. Manolopoulos, i-pi: A python interface for ab initio path integral molecular dynamics simulations, Computer Physics Communications 185 (3) (2014) 1019–1026. 42
2014
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.