Recognition: unknown
Assessing excited-state geometry optimization strategies for adiabatic photophysical energies
Pith reviewed 2026-05-08 04:10 UTC · model grok-4.3
The pith
TDDFT-optimized S1 and T1 geometries produce adiabatic 0-0 energies with mean absolute error below 0.1 eV against experiment.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Adiabatic 0-0 energies evaluated at TDDFT-optimized S1 and T1 geometries show the best agreement with experiment, with a mean absolute error below 0.1 eV. Replacing these geometries with UKS-optimized T1 and ssUKS-optimized S1 structures yields comparable accuracy. Vertical excitation energies are substantially more sensitive to the choice of geometry than the corresponding S1-T1 gaps, which are comparatively more robust because of partial error cancellation.
What carries the argument
Benchmarked comparison of TDDFT, UKS, and ssUKS geometry optimizations, followed by ADC(2) single-point energies plus zero-point vibrational corrections evaluated at those geometries.
If this is right
- S1-T1 gaps remain stable across geometry choices because errors partially cancel.
- UKS and ssUKS optimizations supply an efficient alternative to TDDFT for states with single-determinant character.
- The same geometries remain useful for evaluating singlet-fission energetics in larger molecules such as rubrene.
- Vertical excitation energies change more with geometry choice than the energy gaps between states.
Where Pith is reading between the lines
- These protocols could lower the computational cost of screening candidate molecules for light-harvesting or emissive applications.
- The robustness of the gaps suggests the methods may transfer to other difference properties even when absolute energies are less accurate.
- Testing the same geometry choices with higher-level wavefunction methods would clarify whether the observed accuracy ordering persists beyond ADC(2).
- The findings imply that initial excited-state structures from simpler DFT optimizations can serve as reliable starting points for more expensive calculations.
Load-bearing premise
The chosen molecules and their anion photoelectron spectroscopy data represent general photophysical processes and the excited states possess dominant single-determinant character.
What would settle it
Adiabatic 0-0 energies computed from these optimized geometries that deviate by more than 0.1 eV from new experimental measurements on an expanded set of molecules would disprove the reported accuracy ranking.
Figures
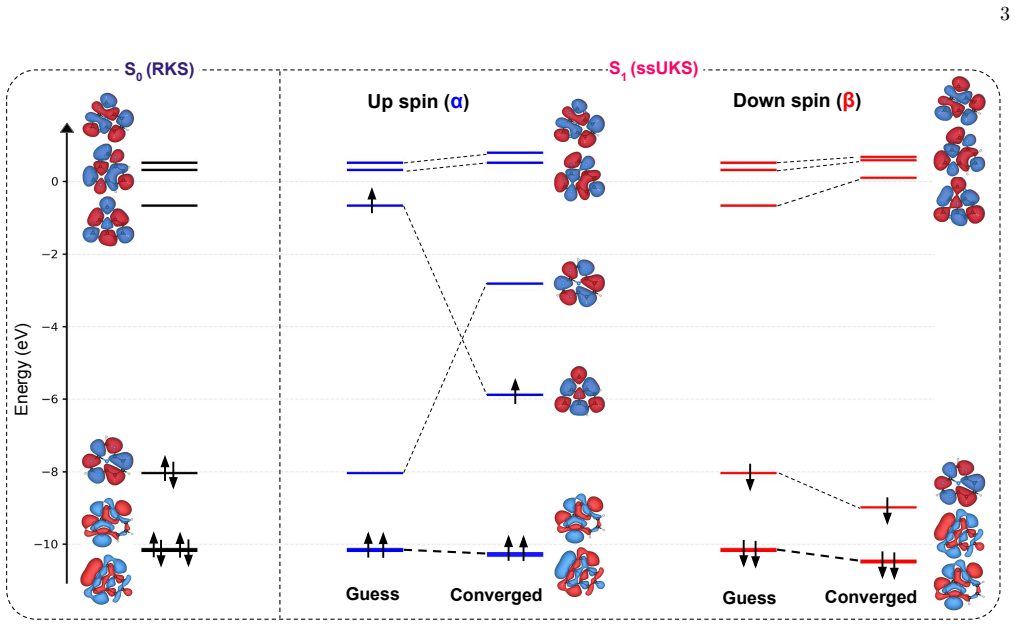



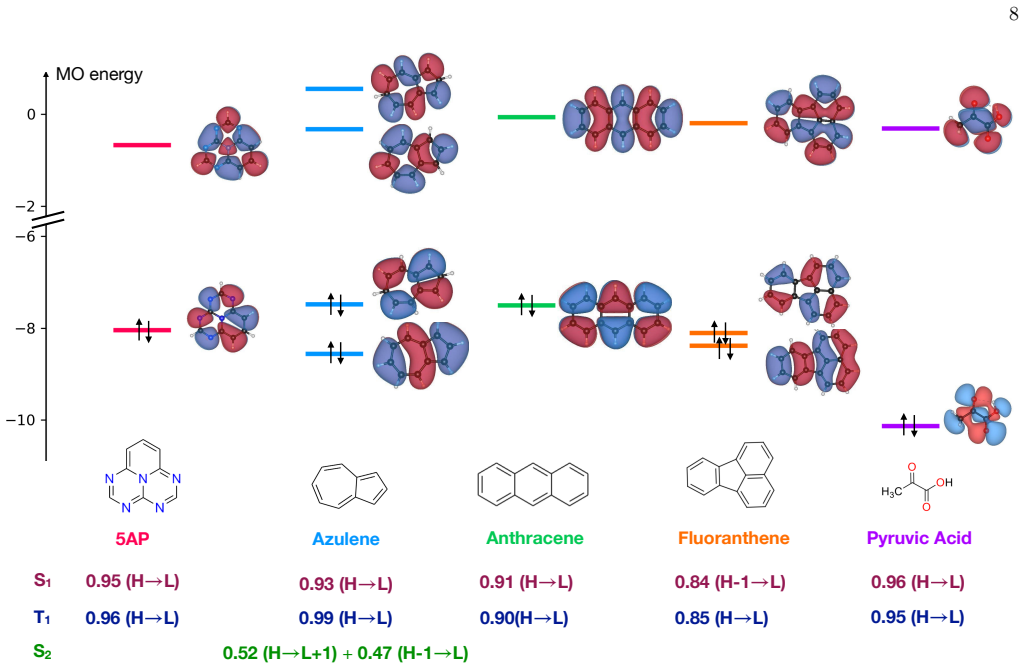

read the original abstract
Accurate prediction of adiabatic $0$-$0$ excited-state energies is crucial for modeling molecular photophysical processes. Here, we benchmark computational strategies for evaluating excited-state energies and singlet-triplet gaps obtained using different geometry-optimization strategies, including time-dependent density functional theory (TDDFT), spin-unrestricted Kohn-Sham (UKS) DFT for triplet states (${\rm T}_1$), and state-specific orbital-optimized UKS (ssUKS) DFT for singlet excited states (${\rm S}_1$). Zero-point vibrational energy corrections are evaluated consistently at the optimized geometries and combined with ADC(2) excitation energies for comparison with experimental anion photoelectron spectroscopy data for a representative set of molecules. Among the protocols considered, adiabatic $0$-$0$ energies evaluated at TDDFT-optimized ${\rm S}_1$ and ${\rm T}_1$ geometries show the best agreement with experiment, with a mean absolute error below 0.1 eV. Replacing these geometries with UKS-optimized ${\rm T}_1$ and ssUKS-optimized ${\rm S}_1$ structures yields comparable accuracy. Vertical excitation energies are substantially more sensitive to the choice of geometry than the corresponding ${\rm S}_1$-${\rm T}_1$ gaps, which are comparatively more robust because of partial error cancellation. As a larger case study, we examine rubrene and find that UKS/ssUKS-based geometries remain useful for evaluating singlet-fission energetics. Overall, UKS/ssUKS-based workflows provide an efficient and accurate route to excited-state geometry optimization and to the evaluation of adiabatic $0$-$0$ energies for states with dominant single-determinant character.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper benchmarks computational strategies for excited-state geometry optimization to obtain adiabatic 0-0 energies and S1-T1 gaps. Protocols using TDDFT, UKS DFT (for T1), and ssUKS DFT (for S1) geometries are evaluated with consistent ZPVE corrections and ADC(2) single-point energies, benchmarked directly against experimental anion photoelectron spectroscopy data for a representative molecular set, plus a rubrene case study on singlet-fission energetics. The central finding is that TDDFT-optimized S1/T1 geometries yield the best experimental agreement (MAE below 0.1 eV), with UKS/ssUKS geometries providing comparable accuracy; vertical energies are more geometry-sensitive than gaps, which benefit from partial error cancellation.
Significance. If the performance claims hold, the work supplies practical, efficiency-oriented guidance for photophysical modeling by showing that orbital-optimized DFT geometries can substitute for TDDFT without substantial loss of accuracy for single-determinant states. The explicit experimental validation, consistent ZPVE treatment, and error-cancellation analysis for gaps are strengths that support broader applicability to processes such as singlet fission.
major comments (2)
- [Computational Methods] Computational Methods section: explicit selection criteria, inclusion/exclusion rules, and the complete list of molecules with references to the anion PES experimental sources are not provided. This information is load-bearing for verifying the robustness of the reported MAE < 0.1 eV and the claim that the set is representative.
- [Results] Results section: statistical uncertainties or error bars on the MAE values for each geometry protocol are not reported. Without them, the assertion that TDDFT geometries are best and that UKS/ssUKS are comparable cannot be assessed for statistical significance.
minor comments (3)
- A table summarizing the benchmark molecules, their experimental 0-0 energies, and the computed values for each protocol would improve readability and reproducibility.
- [Conclusion] The conclusion's caveat on applicability to 'states with dominant single-determinant character' would benefit from a short operational definition or reference to how such character is diagnosed (e.g., via natural orbital occupation numbers).
- Figure legends and axis labels should explicitly name the geometry-optimization method (TDDFT, UKS, ssUKS) rather than using abbreviations alone.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive comments, which have helped us identify areas for improvement. We address each major comment point by point below and outline the revisions we will make.
read point-by-point responses
-
Referee: [Computational Methods] Computational Methods section: explicit selection criteria, inclusion/exclusion rules, and the complete list of molecules with references to the anion PES experimental sources are not provided. This information is load-bearing for verifying the robustness of the reported MAE < 0.1 eV and the claim that the set is representative.
Authors: We agree that these details are essential for full reproducibility and for allowing readers to assess the representativeness of the benchmark set. In the revised manuscript, we will expand the Computational Methods section to explicitly state the selection criteria and inclusion/exclusion rules used to assemble the molecular set. We will also include the complete list of molecules together with the original references to the anion photoelectron spectroscopy experiments from which the adiabatic energies were taken. revision: yes
-
Referee: [Results] Results section: statistical uncertainties or error bars on the MAE values for each geometry protocol are not reported. Without them, the assertion that TDDFT geometries are best and that UKS/ssUKS are comparable cannot be assessed for statistical significance.
Authors: We acknowledge that the absence of statistical uncertainties makes it difficult to judge whether the observed differences in MAE are statistically meaningful. Although the benchmark set is modest in size, we will add the standard deviation of the signed errors and, where appropriate, bootstrap-derived standard errors on the MAE values for each geometry-optimization protocol in the revised Results section. This will allow a quantitative evaluation of the robustness of the conclusion that TDDFT geometries perform best while UKS/ssUKS geometries remain comparable. revision: yes
Circularity Check
No significant circularity identified
full rationale
The paper benchmarks geometry optimization protocols (TDDFT, UKS for T1, ssUKS for S1) by computing adiabatic 0-0 energies and S1-T1 gaps via ADC(2) on top of those geometries, then directly compares the results to independent experimental anion photoelectron spectroscopy data for a representative molecular set. Reported MAEs (<0.1 eV), gap robustness via error cancellation, and the rubrene case study are all external validations; no equations, fitted parameters, or self-citations reduce these quantities to quantities defined inside the paper. The single-determinant caveat is stated explicitly rather than used to force agreement. The derivation chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption ADC(2) provides sufficiently accurate excitation energies when combined with ZPVE corrections at DFT-optimized geometries
- domain assumption The test set of molecules and experimental anion photoelectron data are representative for systems with single-determinant excited states
Reference graph
Works this paper leans on
-
[1]
Wilson, Kenneth D and Styers, William H and Wood, Samuel A and Woods, R Claude and McMahon, Robert J and Liu, Zhe and Yang, Yang and Garand, Etienne , journal=jacs, year=
-
[2]
Vossk. Phys. Chem. Chem. Phys. , volume=. 2015 , url=
2015
-
[3]
2013 , url=
Krylov, Anna I and Gill, Peter MW , journal=. 2013 , url=
2013
-
[4]
2018 , url=
Neese, Frank , journal=WireCMS, volume=. 2018 , url=
2018
-
[5]
2012 , url=
Neese, Frank , journal=WireCMS, volume=. 2012 , url=
2012
-
[6]
2016", note=
M. J. Frisch and others , year="2016", note="
2016
-
[7]
2016 , url=
Gaussian 16. 2016 , url=
2016
-
[8]
2016 , url=
Santoro, Fabrizio and Jacquemin, Denis , journal=WireCMS, volume=. 2016 , url=
2016
-
[9]
2018 , url=
Bremond, Eric and Savarese, Marika and Adamo, Carlo and Jacquemin, Denis , journal=jctc, volume=. 2018 , url=
2018
-
[10]
2018 , url=
Jacquemin, Denis , journal=jctc, volume=. 2018 , url=
2018
-
[11]
2025 , url=
Chanda, Shamik and Saha, Subhasish and Sen, Sangita , journal=jcp, volume=. 2025 , url=
2025
-
[12]
2000 , url=
Tozer, David J and Handy, Nicholas C , journal=pccp, volume=. 2000 , url=
2000
-
[13]
2024 , url=
Kunze, Lukas and Froitzheim, Thomas and Hansen, Andreas and Grimme, Stefan and Mewes, Jan-Michael , journal=jpcl, volume=. 2024 , url=
2024
-
[14]
2025 , url=
Burrow, E Michi and Carmona-Garc. 2025 , url=
2025
-
[15]
2018 , url=
Kregel, Steven J and Thurston, Glen K and Garand, Etienne , journal=jcp, volume=. 2018 , url=
2018
-
[16]
2012 , url=
Jacquemin, Denis and Adamo, Carlo , journal=ijqc, volume=. 2012 , url=
2012
-
[17]
2019 , url=
Loos, Pierre-Fran. 2019 , url=
2019
-
[18]
2009 , url=
Baba, Masaaki and Saitoh, Motohisa and Taguma, Kunio and Shinohara, Keisuke and Yoshida, Kazuto and Semba, Yosuke and Kasahara, Shunji and Nakayama, Naofumi and Goto, Hitoshi and Ishimoto, Takayoshi and others , journal=jcp, volume=. 2009 , url=
2009
-
[19]
1985 , url=
Chan, IY and Dantus, Marcos , journal=jcp, volume=. 1985 , url=
1985
-
[20]
2007 , url=
Melania Oana, C and Krylov, Anna I , journal=jcp, volume=. 2007 , url=
2007
-
[21]
1996 , url=
Bearpark, Michael J and Bernardi, Fernando and Clifford, Simon and Olivucci, Massimo and Robb, Michael A and Smith, Barry R and Vreven, Thom , journal=jacs, volume=. 1996 , url=
1996
-
[22]
1955 , url=
Beer, Michael and Longuet-Higgins, HC , journal=jcp, volume=. 1955 , url=
1955
-
[23]
2023 , url=
Dunlop, David and Ludv. 2023 , url=
2023
-
[24]
2025 , url=
Zheng, Zong-xiang and Wu, Wang-yang and Wan, Hao-bo and Xie, Wen-bin and Yang, Jun and Wang, Zhou and Yang, Lei and Ran, Xue-qin and Xie, Ling-hai , journal=pccp, volume=. 2025 , url=
2025
-
[25]
2022 , url=
Vandaele, Eva and Mali. 2022 , url=
2022
-
[26]
2024 , url=
Tripathy, Vikrant and Flood, Amar H and Raghavachari, Krishnan , journal=jpca, volume=. 2024 , url=
2024
-
[27]
2017 , url=
Tsunoyama, Hironori and Nakajima, Atsushi , journal=. 2017 , url=
2017
-
[28]
2008 , url=
Gilbert, Andrew TB and Besley, Nicholas A and Gill, Peter MW , journal=. 2008 , url=
2008
-
[29]
2025 , url=
Malis, Momir and Luber, Sandra , journal=. 2025 , url=
2025
-
[30]
2023 , url=
Kimber, Patrick and Plasser, Felix , journal=. 2023 , url=
2023
-
[31]
2012 , url=
Neese, Frank , journal=. 2012 , url=
2012
-
[32]
2025 , url=
Neese, Frank , journal=. 2025 , url=
2025
-
[33]
Vahtras, O and Alml. Chem. Phys. Lett. , volume=. 1993 , url=
1993
-
[34]
Kendall, Rick A. and Fr. Theor. Chem. Acc. , volume=. 1997 , url=
1997
-
[35]
, journal=
Yang, Weitao and Ayers, Paul W. , journal=. 2024 , url=
2024
-
[36]
2021 , publisher=
Hait, Diptarka and Head-Gordon, Martin , journal=. 2021 , publisher=
2021
-
[37]
2013 , publisher=
Smith, Millicent B and Michl, Josef , journal=. 2013 , publisher=
2013
-
[38]
Chen, Xian-Kai and Kim, Dongwook and Br. Acc. Chem. Res. , volume=. 2018 , url=
2018
-
[39]
2012 , url=
Uoyama, Hiroki and Goushi, Kenichi and Shizu, Katsuyuki and Nomura, Hiroko and Adachi, Chihaya , journal=. 2012 , url=
2012
-
[40]
2022 , url=
Aizawa, Naoya and Pu, Yong-Jin and Harabuchi, Yu and Nihonyanagi, Atsuko and Ibuka, Ryotaro and Inuzuka, Hiroyuki and Dhara, Barun and Koyama, Yuki and Nakayama, Ken-ichi and Maeda, Satoshi and others , journal=. 2022 , url=
2022
-
[41]
2023 , url=
Won, Taehyun and Nakayama, Ken-ichi and Aizawa, Naoya , journal=cprev, volume=. 2023 , url=
2023
-
[42]
2022 , url=
Li, Jie and Li, Zhi and Liu, Hui and Gong, Heqi and Zhang, Jincheng and Yao, Yali and Guo, Qiang , journal=. 2022 , url=
2022
-
[43]
2019 , url=
de Silva, Piotr , journal=. 2019 , url=
2019
-
[44]
2020 , publisher=
Veys, Koen and Escudero, Daniel , journal=jpca, volume=. 2020 , publisher=
2020
-
[45]
2014 , publisher=
Fang, Changfeng and Oruganti, Baswanth and Durbeej, Bo , journal=jpca, volume=. 2014 , publisher=
2014
-
[46]
2025 , publisher=
Sinyavskiy, Andrey and Malis, Momir and Luber, Sandra , journal=. 2025 , publisher=
2025
-
[47]
2026 , url =
Vigneshwaran, V and Basumatary, Swrangsar and Beypi, Chitralekha and Akshay, CP and Ghosh, Soumen , journal=. 2026 , url =
2026
-
[48]
2008 , publisher=
Cheng, Chiao-Lun and Wu, Qin and Van Voorhis, Troy , journal=. 2008 , publisher=
2008
-
[49]
2019 , url=
Ehrmaier, Johannes and Rabe, Emily J and Pristash, Sarah R and Corp, Kathryn L and Schlenker, Cody W and Sobolewski, Andrzej L and Domcke, Wolfgang , journal=jpca, volume=. 2019 , url=
2019
-
[50]
2015 , url=
Kondakov, Denis Y , journal=. 2015 , url=
2015
-
[51]
2012 , url=
Peach, Michael JG and Tozer, David J , journal=jpca, volume=. 2012 , url=
2012
-
[52]
1997 , url=
Kendall, Rick A and Fr. 1997 , url=
1997
-
[53]
2017 , url=
Sutton, Christopher and Tummala, Naga Rajesh and Beljonne, David and Brédas, Jean-Luc , journal=. 2017 , url=
2017
-
[54]
2012 , url=
Ma, Lin and Zhang, Keke and Kloc, Christian and Sun, Handong and Michel-Beyerle, Maria E and Gurzadyan, Gagik G , journal=pccp, volume=. 2012 , url=
2012
-
[55]
2023 , url=
Loos, Pierre-Fran. 2023 , url=
2023
-
[56]
1990 , url=
Jensen, Frank , journal=cpl, volume=. 1990 , url=
1990
-
[57]
2022 , url=
Froitzheim, Thomas and Grimme, Stefan and Mewes, Jan-Michael , journal=jctc, volume=. 2022 , url=
2022
-
[58]
2016 , url=
Hait, Diptarka and Zhu, Tianyu and McMahon, David P and Van Voorhis, Troy , journal=jctc, volume=. 2016 , url=
2016
-
[59]
2020 , url=
Hait, Diptarka and Head-Gordon, Martin , journal=jctc, volume=. 2020 , url=
2020
-
[60]
2025 , url=
Majumdar, Atreyee and Das, Surajit and Ramakrishnan, Raghunathan , journal=. 2025 , url=
2025
-
[61]
2025 , url=
Majumdar, Atreyee and Ramakrishnan, Raghunathan , journal=cej, pages=. 2025 , url=
2025
-
[62]
2025 , url=
Jindal, Komal and Majumdar, Atreyee and Ramakrishnan, Raghunathan , journal=pccp, volume=. 2025 , url=
2025
-
[63]
1977 , url=
Ziegler, Tom and Rauk, Arvi and Baerends, Evert J , journal=tca, volume=. 1977 , url=
1977
-
[64]
2024 , url=
Pino-Rios, Ricardo and B. 2024 , url=
2024
-
[65]
and Knott, W
Schiedt, J. and Knott, W. J. and Le Barbu, K. and Schlag, E. W. and Weinkauf, R. , journal=jcp, volume=. 2000 , doi=
2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.