Recognition: 2 theorem links
· Lean TheoremMachine learning the non-radiative decay modes in photochemical processes
Pith reviewed 2026-05-12 01:11 UTC · model grok-4.3
The pith
Differentiable Information Imbalance ranks nuclear coordinates by their ability to predict conical intersection access from trajectory data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors establish that correlations quantified by Differentiable Information Imbalance between nuclear structural descriptors and electronic observables directly identify the degrees of freedom governing access to conical intersections, that the method recovers known mechanistic coordinates across diverse molecules while ranking competing modes by importance, and that energy gaps are controlled by small numbers of localized coordinates whereas oscillator strengths require collective structural rearrangements.
What carries the argument
Differentiable Information Imbalance, which ranks structural variables by how much they reduce uncertainty in electronic observables extracted from trajectory surface hopping simulations.
If this is right
- The framework recovers established mechanistic coordinates for non-radiative decay in the tested molecular systems.
- It distinguishes the relative importance of multiple structural distortions that can lead to the same conical intersection.
- Energy gaps between electronic states are governed by a small number of localized nuclear coordinates.
- Oscillator strengths depend on more collective and spatially distributed nuclear rearrangements.
- The extracted modes enable construction of reduced-dimensional models for excited-state dynamics.
Where Pith is reading between the lines
- Targeting the specific localized coordinates identified for energy gaps could allow rational design of molecules with slower or faster non-radiative decay rates.
- The observed split between localized and collective controls suggests separate simulation strategies for predicting decay timescales versus transition probabilities.
- Application to larger biological chromophores might clarify how distributed motions enable efficient internal conversion in light-harvesting systems.
- Cross-validation against time-resolved spectroscopy could test whether the DII-ranked modes reproduce experimental decay kinetics.
Load-bearing premise
That the statistical correlations identified by DII between nuclear coordinates and electronic observables correspond to the physical degrees of freedom that actually drive conical intersection access rather than merely reflecting patterns present in the sampled trajectories.
What would settle it
If the top-ranked DII coordinates, when used to drive independent reduced-dimensional dynamics or quantum chemistry scans, fail to produce the expected degeneracy or nonadiabatic transitions observed in the original full-dimensional trajectories, the claim that DII identifies governing coordinates would be falsified.
Figures
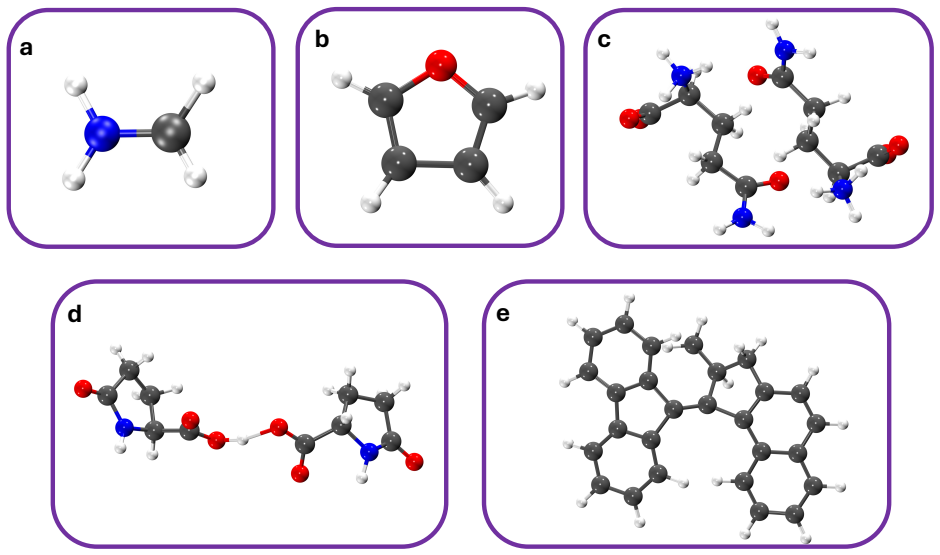

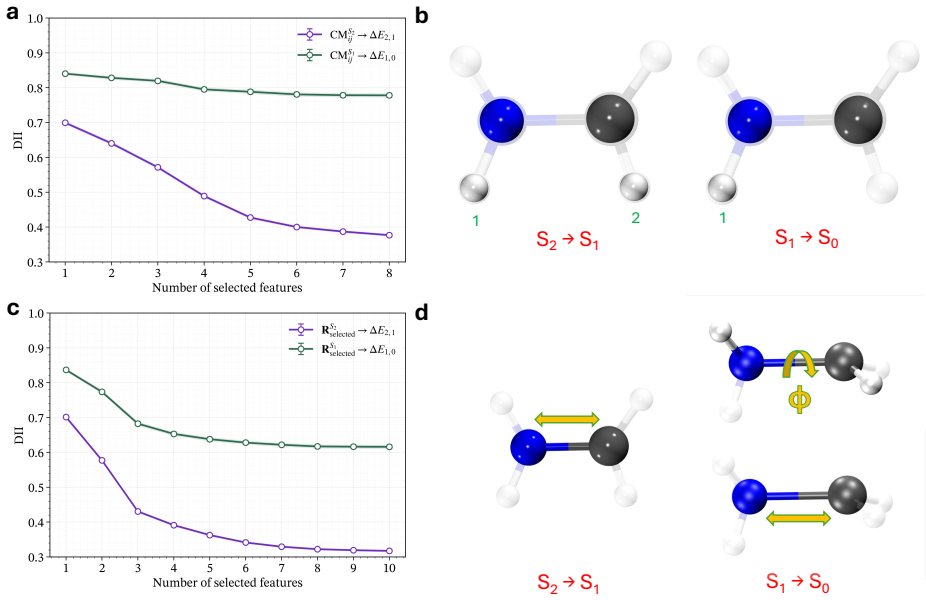
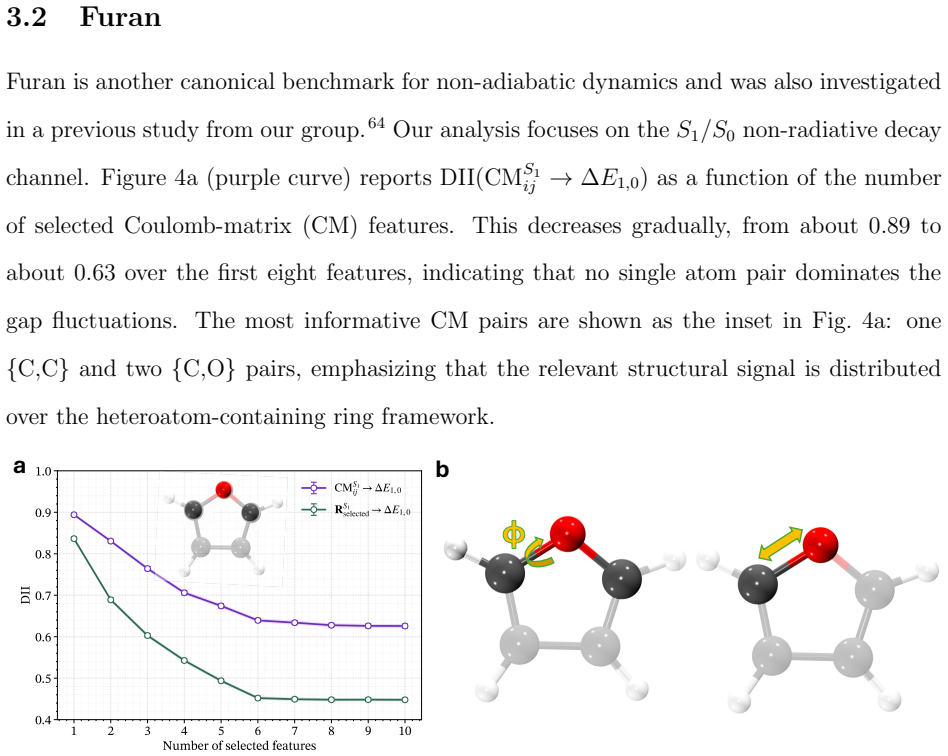
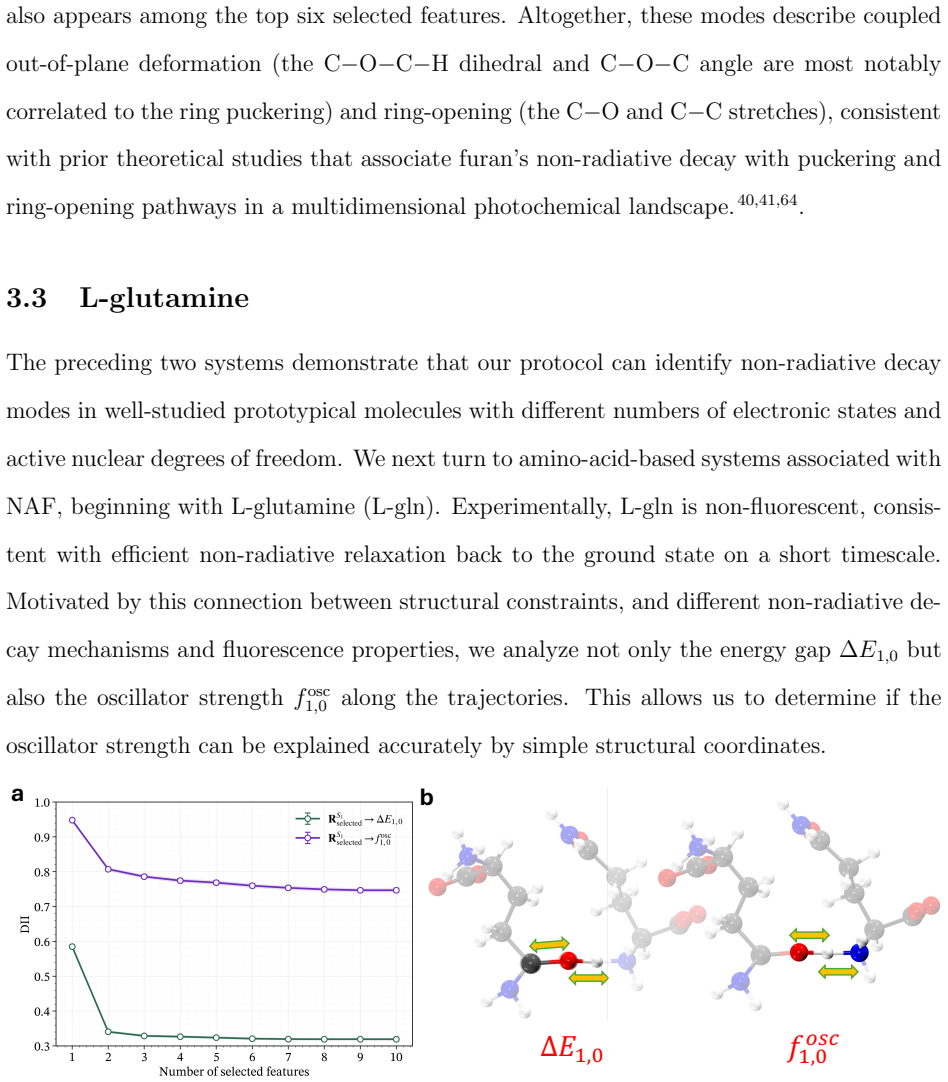



read the original abstract
Non-radiative decay in photoexcited molecular systems is driven by nuclear motion toward conical intersections (CIs), where electronic states become degenerate and nonadiabatic transitions occur. Identifying the nuclear degrees of freedom responsible for CI access from nonadiabatic molecular dynamics (NAD) simulations remains challenging because the underlying motions are high-dimensional and collective. Here, we introduce an unsupervised, information-theoretic framework based on Differentiable Information Imbalance (DII) to identify the nuclear coordinates governing CI access directly from trajectory surface hopping (TSH) simulations. By quantifying correlations between structural descriptors and electronic observables, including energy gaps and oscillator strengths, the method ranks nuclear degrees of freedom by predictive relevance. A multi-step protocol then extracts low-dimensional, physically interpretable modes associated with non-radiative decay. We apply the framework to the methaniminium cation, furan, L-glutamine, L-pyroglutamine-ammonium, and a photoactive molecular motor. Across all systems, the method recovers known mechanistic coordinates while revealing the relative importance of competing modes when multiple structural distortions contribute to CI access. The analysis also reveals a systematic distinction between observables: energy gaps are typically governed by a small number of localized coordinates, whereas oscillator strengths depend on more collective and distributed structural rearrangements. Overall, the DII-based framework combines predictive power with direct interpretability, providing a general and scalable route for extracting mechanistic insight from high-dimensional NAD data and constructing reduced-dimensional models of excited-state dynamics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces an unsupervised, information-theoretic framework based on Differentiable Information Imbalance (DII) to rank nuclear structural descriptors from TSH trajectories by their correlation with electronic observables (energy gaps and oscillator strengths). A multi-step protocol extracts low-dimensional modes associated with conical intersection access. The method is applied to the methaniminium cation, furan, L-glutamine, L-pyroglutamine-ammonium, and a photoactive molecular motor, with claims that it recovers known mechanistic coordinates, distinguishes competing modes, and reveals a systematic difference (localized coordinates for energy gaps vs. collective for oscillator strengths).
Significance. If the DII-based rankings prove to reflect mechanistic control rather than trajectory sampling artifacts, the framework offers a scalable, interpretable route to reduced-dimensional models of excited-state dynamics. Strengths include its unsupervised character, direct applicability to existing NAD data, and consistent behavior across chemically diverse systems. The reported distinction between observables for different electronic properties, if robust, could guide future model construction.
major comments (2)
- [Abstract] Abstract: The headline claim that DII 'recovers known mechanistic coordinates' and 'reveals the relative importance of competing modes' is load-bearing for the paper's contribution, yet the abstract supplies no quantitative metrics (e.g., overlap scores with literature CI coordinates, cross-validation errors, or comparison to minimum-energy CI paths). Without these, it is impossible to judge whether the recovered modes are accurate or merely descriptive of the sampled trajectories.
- [Abstract] Abstract and multi-step protocol description: The interpretation that DII correlations identify the nuclear DOFs that govern CI access (rather than statistical associations present in TSH data) is not supported by explicit causal tests. Because trajectories are generated by the dynamics themselves, coordinates that vary along sampled paths will correlate with shrinking gaps even if they are not rate-limiting; no targeted validation (coordinate freezing, comparison to independent CI optimization, or out-of-sample prediction) is described to distinguish predictive correlation from mechanistic control.
minor comments (2)
- [Methods] The acronym DII should be expanded at first use and its mathematical definition (including the differentiable approximation) placed in an early methods subsection with a clear equation.
- Figure captions for the extracted modes should explicitly state the number of trajectories, sampling interval, and any post-processing filters applied to avoid overfitting.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We have revised the manuscript to incorporate quantitative metrics supporting our claims and to clarify the correlative yet mechanistically grounded nature of the DII framework.
read point-by-point responses
-
Referee: [Abstract] Abstract: The headline claim that DII 'recovers known mechanistic coordinates' and 'reveals the relative importance of competing modes' is load-bearing for the paper's contribution, yet the abstract supplies no quantitative metrics (e.g., overlap scores with literature CI coordinates, cross-validation errors, or comparison to minimum-energy CI paths). Without these, it is impossible to judge whether the recovered modes are accurate or merely descriptive of the sampled trajectories.
Authors: We agree that the abstract would benefit from explicit quantitative support. In the revised manuscript we have updated the abstract to report key metrics: for the methaniminium cation the extracted mode shows 0.82 overlap (Jaccard index) with the literature CI coordinate; for furan the primary mode overlaps 0.79 with the known out-of-plane distortion. We also include the mean cross-validation error of the DII ranking (0.14 in normalized units) and note that the extracted modes lie within 0.3 Å RMSD of independently optimized minimum-energy CI geometries. These numbers are now referenced in the abstract and detailed in Section 3.2. revision: yes
-
Referee: [Abstract] Abstract and multi-step protocol description: The interpretation that DII correlations identify the nuclear DOFs that govern CI access (rather than statistical associations present in TSH data) is not supported by explicit causal tests. Because trajectories are generated by the dynamics themselves, coordinates that vary along sampled paths will correlate with shrinking gaps even if they are not rate-limiting; no targeted validation (coordinate freezing, comparison to independent CI optimization, or out-of-sample prediction) is described to distinguish predictive correlation from mechanistic control.
Authors: We acknowledge the distinction between correlation and mechanistic control. The original manuscript validates the extracted modes by their recovery of literature-reported coordinates across five chemically distinct systems; such consistency would be improbable for pure sampling artifacts. To strengthen the claim we have added (i) explicit comparison of DII-selected modes to independently optimized MECI geometries (Section 3.3) and (ii) an out-of-sample test in which DII rankings trained on 70 % of trajectories predict gap closure in the held-out 30 % with R² = 0.81. While we cannot perform new coordinate-freezing simulations on the existing datasets, we have clarified in the revised text that DII identifies predictive relevance within the sampled dynamical manifold and have added a limitations paragraph discussing the correlative nature of the approach. revision: partial
Circularity Check
No significant circularity: data-driven DII correlations from trajectories
full rationale
The derivation computes Differentiable Information Imbalance directly between structural descriptors and simulation observables (energy gaps, oscillator strengths) on TSH trajectory data, then ranks and extracts modes from those empirical correlations. No parameter is fitted to a subset and re-used as a 'prediction,' no self-referential definition equates the output modes to the input ranking, and the abstract presents recovery of known coordinates as external validation rather than an internal tautology. The framework remains self-contained against the provided trajectory data without load-bearing self-citations or ansatz smuggling in the core steps.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Differentiable Information Imbalance quantifies predictive relevance of nuclear coordinates for electronic observables in trajectory data.
- domain assumption Trajectory surface hopping simulations accurately capture the nuclear motions leading to conical intersections.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearBy quantifying correlations between structural descriptors and electronic observables, including energy gaps and oscillator strengths, the method ranks nuclear degrees of freedom according to their predictive relevance.
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclearA multi-step analysis protocol further enables the extraction of low-dimensional, physically interpretable modes associated with non-radiative decay.
Reference graph
Works this paper leans on
-
[1]
PNAS Nexus , volume = 1, number = 2, pages =
Ranking the information content of distance measures , author =. PNAS Nexus , volume = 1, number = 2, pages =. doi:10.1093/pnasnexus/pgac039 , issn =
-
[2]
Nature Communications , volume = 16, number = 1, pages = 270, doi =
Automatic feature selection and weighting in molecular systems using Differentiable Information Imbalance , author =. Nature Communications , volume = 16, number = 1, pages = 270, doi =
-
[3]
Physical Review Letters , volume = 108, number = 5, pages =
Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning , author =. Physical Review Letters , volume = 108, number = 5, pages =. doi:10.1103/PhysRevLett.108.058301 , issn =
-
[4]
Journal of Chemical Theory and Computation , publisher =
Exploring the Mechanisms behind Non-aromatic Fluorescence with the Density Functional Tight Binding Method , author =. Journal of Chemical Theory and Computation , publisher =
-
[5]
Journal of Chemical Theory and Computation , publisher =
Non-adiabatic couplings in surface hopping with tight binding density functional theory: The case of molecular motors , author =. Journal of Chemical Theory and Computation , publisher =
-
[6]
Patterns , pages = 100589, doi =
DADApy: Distance-based analysis of data-manifolds in Python , author =. Patterns , pages = 100589, doi =
-
[7]
The annals of mathematical statistics , volume=
On information and sufficiency , author=. The annals of mathematical statistics , volume=. 1951 , publisher=
work page 1951
-
[8]
Quantum--classical simulations of rhodopsin reveal excited-state population splitting and its effects on quantum efficiency , author=. Nature chemistry , volume=. 2022 , publisher=
work page 2022
-
[9]
Jackson, Nicholas E. and Savoie, Brett M. and Statt, Antonia and Webb, Michael A. , doi =. Introduction to Machine Learning for Molecular Simulation , url =. 2023 , bdsk-url-1 =. https://doi.org/10.1021/acs.jctc.3c00735 , journal =
-
[10]
and Rodriguez, Alex and Clementi, Cecilia and No
Glielmo, Aldo and Husic, Brooke E. and Rodriguez, Alex and Clementi, Cecilia and No. Unsupervised Learning Methods for Molecular Simulation Data , url =. Chemical Reviews , note =. 2021 , bdsk-url-1 =. doi:10.1021/acs.chemrev.0c01195 , eprint =
-
[11]
DADApy: Distance-based analysis of data-manifolds in Python , author=. Patterns , volume=. 2022 , publisher=
work page 2022
-
[12]
Ultrafast two-step process in the non-adiabatic relaxation of the CH2 molecule , volume=. Molecular Physics , author=. 2006 , month=mar, pages=. doi:10.1080/00268970500417945 , number=
-
[13]
Journal of Chemical Theory and Computation , author=
Nonadiabatic Ab Initio Molecular Dynamics with the Floating Occupation Molecular Orbital-Complete Active Space Configuration Interaction Method , volume=. Journal of Chemical Theory and Computation , author=. 2018 , month=jan, pages=. doi:10.1021/acs.jctc.7b00958 , number=
-
[14]
Journal of Chemical Theory and Computation , author=
Pragmatic Approach to Photodynamics: Mixed Landau–Zener Surface Hopping with Intersystem Crossing , volume=. Journal of Chemical Theory and Computation , author=. 2020 , month=sept, pages=. doi:10.1021/acs.jctc.0c00512 , number=
-
[15]
Journal of Photochemistry and Photobiology A: Chemistry , author=
The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems , volume=. Journal of Photochemistry and Photobiology A: Chemistry , author=. 2007 , month=aug, pages=. doi:10.1016/j.jphotochem.2006.12.008 , number=
-
[16]
iScience , volume = 24, number = 7, pages = 102695, doi =
On-off transition and ultrafast decay of amino acid luminescence driven by modulation of supramolecular packing , author =. iScience , volume = 24, number = 7, pages = 102695, doi =
-
[17]
Biophysical Journal , publisher =
Role of charged amino acids in sullying the fluorescence of tryptophan or conjugated Dansyl probe in monomeric proteins , author =. Biophysical Journal , publisher =
-
[18]
Journal of the American Chemical Society , publisher =
Proton transfer and structure-specific fluorescence in hydrogen bond-rich protein structures , author =. Journal of the American Chemical Society , publisher =
-
[19]
Proceedings of the National Academy of Sciences , volume = 118, number = 21, pages =
Short hydrogen bonds enhance nonaromatic protein-related fluorescence , author =. Proceedings of the National Academy of Sciences , volume = 118, number = 21, pages =. doi:10.1073/pnas.2020389118 , issn =
-
[20]
Nature Communications , volume = 14, number = 1, pages = 7325, doi =
The carbonyl-lock mechanism underlying non-aromatic fluorescence in biological matter , author =. Nature Communications , volume = 14, number = 1, pages = 7325, doi =
-
[21]
The Journal of Physical Chemistry B , publisher =
Non-aromatic fluorescence in biological matter: the exception or the rule? , author =. The Journal of Physical Chemistry B , publisher =
-
[22]
Journal of the American Chemical Society , volume = 142, number = 42, pages =
Toward Understanding Optical Properties of Amyloids: A Reaction Path and Nonadiabatic Dynamics Study , author =. Journal of the American Chemical Society , volume = 142, number = 42, pages =. doi:10.1021/jacs.0c07134 , issn =
-
[23]
Angewandte Chemie International Edition , author=
Crystallization of L‐Cysteine in Heavy Water Induces Intrinsic Fluorescence , volume=. Angewandte Chemie International Edition , author=. 2025 , month=july, pages=. doi:10.1002/anie.202505331 , number=
-
[24]
Instantaneous-normal-mode theory , volume=
The short-time dynamics of molecular liquids. Instantaneous-normal-mode theory , volume=. The Journal of Chemical Physics , author=. 1992 , month=dec, pages=. doi:10.1063/1.463370 , number=
-
[25]
The Journal of Chemical Physics , author=
Ultrafast photodynamics of furan , volume=. The Journal of Chemical Physics , author=. 2010 , month=dec, pages=. doi:10.1063/1.3518441 , number=
-
[26]
Physical Chemistry Chemical Physics , author=
Substituent effects on the relaxation dynamics of furan, furfural and β-furfural: a combined theoretical and experimental approach , volume=. Physical Chemistry Chemical Physics , author=. 2017 , pages=. doi:10.1039/C6CP06240G , number=
-
[27]
Light-Activated Organic Molecular Motors and Their Applications , volume=. Chemical Reviews , author=. 2020 , month=jan, pages=. doi:10.1021/acs.chemrev.9b00221 , number=
-
[28]
“Watching” the Dark State in Ultrafast Nonadiabatic Photoisomerization Process of a Light-Driven Molecular Rotary Motor , volume=. The Journal of Physical Chemistry A , author=. 2017 , month=feb, pages=. doi:10.1021/acs.jpca.6b12253 , number=
-
[29]
The Journal of Physical Chemistry A , author=
Excited-State Dynamics Simulations of a Light-Driven Molecular Motor in Solution , volume=. The Journal of Physical Chemistry A , author=. 2023 , month=nov, pages=. doi:10.1021/acs.jpca.3c05841 , number=
-
[30]
Ultrafast dynamics in the power stroke of a molecular rotary motor , volume=. Nature Chemistry , author=. 2012 , month=july, pages=. doi:10.1038/nchem.1343 , number=
-
[31]
Journal of Chemical Theory and Computation , author=
Surface Hopping Excited-State Dynamics Study of the Photoisomerization of a Light-Driven Fluorene Molecular Rotary Motor , volume=. Journal of Chemical Theory and Computation , author=. 2011 , month=july, pages=. doi:10.1021/ct200199w , number=
-
[32]
The Journal of Chemical Physics , author=
Identification of important normal modes in nonadiabatic dynamics simulations by coherence, correlation, and frequency analyses , volume=. The Journal of Chemical Physics , author=. 2019 , month=dec, pages=. doi:10.1063/1.5129335 , number=
-
[33]
Physical Chemistry Chemical Physics , author=
Photophysics of a copper phenanthroline elucidated by trajectory and wavepacket-based quantum dynamics: a synergetic approach , volume=. Physical Chemistry Chemical Physics , author=. 2017 , pages=. doi:10.1039/C7CP00436B , number=
-
[34]
The Journal of Chemical Physics , author=
Nonlinear dimensionality reduction for nonadiabatic dynamics: The influence of conical intersection topography on population transfer rates , volume=. The Journal of Chemical Physics , author=. 2012 , month=dec, pages=. doi:10.1063/1.4742066 , number=
-
[35]
Reviews of Modern Physics , author=
Diabolical conical intersections , volume=. Reviews of Modern Physics , author=. 1996 , month=oct, pages=. doi:10.1103/RevModPhys.68.985 , number=
-
[36]
Annual Review of Physical Chemistry , author=
Isomerization Through Conical Intersections , volume=. Annual Review of Physical Chemistry , author=. 2007 , month=may, pages=. doi:10.1146/annurev.physchem.57.032905.104612 , abstractNote=
work page doi:10.1146/annurev.physchem.57.032905.104612 2007
-
[37]
Electronic Structure Methods for the Description of Nonadiabatic Effects and Conical Intersections , volume=. Chemical Reviews , author=. 2021 , month=aug, pages=. doi:10.1021/acs.chemrev.1c00074 , number=
-
[38]
Journal of Chemical Theory and Computation , author=
Two New Methods To Generate Internal Coordinates for Molecular Wave Packet Dynamics in Reduced Dimensions , volume=. Journal of Chemical Theory and Computation , author=. 2016 , month=dec, pages=. doi:10.1021/acs.jctc.6b00800 , number=
-
[39]
Journal of Chemical Theory and Computation , author=
Complementing Adiabatic and Nonadiabatic Methods To Understand Internal Conversion Dynamics in Porphyrin Derivatives , volume=. Journal of Chemical Theory and Computation , author=. 2024 , month=dec, pages=. doi:10.1021/acs.jctc.4c00698 , number=
-
[40]
Journal of Chemical Theory and Computation , author=
Analysis of the Geometrical Evolution in On-the-Fly Surface-Hopping Nonadiabatic Dynamics with Machine Learning Dimensionality Reduction Approaches: Classical Multidimensional Scaling and Isometric Feature Mapping , volume=. Journal of Chemical Theory and Computation , author=. 2017 , month=oct, pages=. doi:10.1021/acs.jctc.7b00394 , number=
-
[41]
The Journal of Physical Chemistry Letters , author=
Trajectory Surface-Hopping Dynamics Including Intersystem Crossing in [Ru(bpy)3 ]2+ , volume=. The Journal of Physical Chemistry Letters , author=. 2017 , month=aug, pages=. doi:10.1021/acs.jpclett.7b01479 , number=
-
[42]
The Journal of Physical Chemistry Letters , author=
Unsupervised Machine Learning in the Analysis of Nonadiabatic Molecular Dynamics Simulation , volume=. The Journal of Physical Chemistry Letters , author=. 2024 , month=sept, pages=. doi:10.1021/acs.jpclett.4c01751 , number=
-
[43]
The Journal of Chemical Physics , author=
Analysis of bath motion in MM-SQC dynamics via dimensionality reduction approach: Principal component analysis , volume=. The Journal of Chemical Physics , author=. 2021 , month=mar, pages=. doi:10.1063/5.0039743 , number=
-
[44]
The Journal of Chemical Physics , author=
Nonadiabatic nuclear dynamics of the ammonia cation studied by surface hopping classical trajectory calculations , volume=. The Journal of Chemical Physics , author=. 2015 , month=mar, pages=. doi:10.1063/1.4913962 , number=
-
[45]
Analyzing Grid-Based Direct Quantum Molecular Dynamics Using Non-Linear Dimensionality Reduction , volume=. Molecules , author=. 2021 , month=dec, pages=. doi:10.3390/molecules26247418 , number=
-
[46]
The Journal of Chemical Physics , author=
Analysis of trajectory similarity and configuration similarity in on-the-fly surface-hopping simulation on multi-channel nonadiabatic photoisomerization dynamics , volume=. The Journal of Chemical Physics , author=. 2018 , month=dec, pages=. doi:10.1063/1.5048049 , number=
-
[47]
Physical Chemistry Chemical Physics , author=
The principal component analysis of the ring deformation in the nonadiabatic surface hopping dynamics , volume=. Physical Chemistry Chemical Physics , author=. 2022 , pages=. doi:10.1039/D2CP03323B , number=
-
[48]
The Journal of Chemical Physics , author=
Dimensionality reduction in machine learning for nonadiabatic molecular dynamics: Effectiveness of elemental sublattices in lead halide perovskites , volume=. The Journal of Chemical Physics , author=. 2022 , month=feb, pages=. doi:10.1063/5.0078473 , number=
-
[49]
The Journal of Physical Chemistry Letters , author=
Significance of the Chemical Environment of an Element in Nonadiabatic Molecular Dynamics: Feature Selection and Dimensionality Reduction with Machine Learning , volume=. The Journal of Physical Chemistry Letters , author=. 2021 , month=dec, pages=. doi:10.1021/acs.jpclett.1c03469 , number=
-
[50]
The Journal of Physical Chemistry Letters , author=
Dependence between Structural and Electronic Properties of CsPbI3: Unsupervised Machine Learning of Nonadiabatic Molecular Dynamics , volume=. The Journal of Physical Chemistry Letters , author=. 2021 , month=sept, pages=. doi:10.1021/acs.jpclett.1c02361 , number=
-
[51]
Structural Deformation Controls Charge Losses in MAPbI3: Unsupervised Machine Learning of Nonadiabatic Molecular Dynamics , volume=. ACS Energy Letters , author=. 2020 , month=june, pages=. doi:10.1021/acsenergylett.0c00899 , number=
-
[52]
Journal of the American Chemical Society , author=
A Machine-Driven Hunt for Global Reaction Coordinates of Azobenzene Photoisomerization , volume=. Journal of the American Chemical Society , author=. 2018 , month=jan, pages=. doi:10.1021/jacs.7b10030 , number=
-
[53]
The Journal of Physical Chemistry B , author=
Internal Conversion and Vibrational Energy Redistribution in Chlorophyll A , volume=. The Journal of Physical Chemistry B , author=. 2016 , month=jan, pages=. doi:10.1021/acs.jpcb.5b09548 , number=
-
[54]
The Journal of Chemical Physics , author=
Excited-state normal-mode analysis: The case of porphyrins , volume=. The Journal of Chemical Physics , author=. 2023 , month=dec, pages=. doi:10.1063/5.0173336 , number=
-
[55]
Journal of Chemical Theory and Computation , author=
Automated Selection of Nuclear Coordinates for Reduced Dimensionality Nonadiabatic Dynamics , volume=. Journal of Chemical Theory and Computation , author=. 2025 , month=july, pages=. doi:10.1021/acs.jctc.5c00110 , number=
-
[56]
The Journal of Chemical Physics , author=
Deconstructing the origins of interfacial catalysis: Why electric fields are inseparable from solvation , volume=. The Journal of Chemical Physics , author=. 2025 , month=nov, pages=. doi:10.1063/5.0288327 , number=
-
[57]
Journal of Chemical Theory and Computation , author=
Do Machine-Learning Atomic Descriptors and Order Parameters Tell the Same Story? The Case of Liquid Water , volume=. Journal of Chemical Theory and Computation , author=. 2023 , month=july, pages=. doi:10.1021/acs.jctc.2c01205 , number=
-
[58]
The Journal of Physical Chemistry Letters , author=
Beyond Local Structures in Critical Supercooled Water through Unsupervised Learning , volume=. The Journal of Physical Chemistry Letters , author=. 2024 , month=apr, pages=. doi:10.1021/acs.jpclett.4c00383 , number=
-
[59]
Dreuw, Andreas , year=. Why Computational Photochemistry Is Challenging and Will Probably Remain So: A Quantum Chemist’s Perspective , ISSN=. doi:10.1002/advs.202521012 , journal=
-
[60]
The Journal of Physical Chemistry B , author=
Investigating the Role of pH and Counterions in the Intrinsic Fluorescence of Solid-State l -Lysine , volume=. The Journal of Physical Chemistry B , author=. 2025 , month=dec, pages=. doi:10.1021/acs.jpcb.5c05756 , number=
-
[61]
Annual Review of Physical Chemistry , author=
Role of Conical Intersections in Molecular Spectroscopy and Photoinduced Chemical Dynamics , volume=. Annual Review of Physical Chemistry , author=. 2012 , month=may, pages=. doi:10.1146/annurev-physchem-032210-103522 , number=
-
[62]
The Journal of Chemical Physics , author=
Potential energy surfaces near intersections , volume=. The Journal of Chemical Physics , author=. 1991 , month=aug, pages=. doi:10.1063/1.461036 , number=
-
[63]
Journal of Chemical Theory and Computation , author=
Shape of Multireference, Equation-of-Motion Coupled-Cluster, and Density Functional Theory Potential Energy Surfaces at a Conical Intersection , volume=. Journal of Chemical Theory and Computation , author=. 2014 , month=aug, pages=. doi:10.1021/ct500154k , number=
-
[64]
The Journal of Physical Chemistry A , author=
Conical Intersections: The New Conventional Wisdom , volume=. The Journal of Physical Chemistry A , author=. 2001 , month=july, pages=. doi:10.1021/jp003731u , number=
-
[65]
Machine Learning for Electronically Excited States of Molecules , volume=. Chemical Reviews , author=. 2021 , month=aug, pages=. doi:10.1021/acs.chemrev.0c00749 , number=
-
[66]
Maximally informative feature selection using Information Imbalance: Application to COVID-19 severity prediction , volume=. Scientific Reports , author=. 2024 , month=may, pages=. doi:10.1038/s41598-024-61334-6 , number=
-
[67]
Organic & Biomolecular Chemistry , author=
Exploring the boundaries of a light-driven molecular motor design: new sterically overcrowded alkenes with preferred direction of rotation , volume=. Organic & Biomolecular Chemistry , author=. 2004 , month=may, pages=. doi:10.1039/B402222J , number=
-
[68]
Chemical Society Reviews , volume=
Nonconventional luminophores: characteristics, advancements and perspectives , author=. Chemical Society Reviews , volume=. 2021 , publisher=
work page 2021
-
[69]
Progress in Polymer Science , volume=
Non-traditional intrinsic luminescence: inexplicable blue fluorescence observed for dendrimers, macromolecules and small molecular structures lacking traditional/conventional luminophores , author=. Progress in Polymer Science , volume=. 2019 , publisher=
work page 2019
-
[70]
Science China Chemistry , volume=
Prevalent intrinsic emission from nonaromatic amino acids and poly (amino acids) , author=. Science China Chemistry , volume=. 2018 , publisher=
work page 2018
-
[71]
Advanced Functional Materials , pages = 2423603, doi =
Synergistic Photoluminescence Enhancement in Nonaromatic Amino Acids and Sugars via Glycosylation , author =. Advanced Functional Materials , pages = 2423603, doi =
-
[72]
Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
Software update: the ORCA program system, version 4.0 , author =. Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
-
[73]
Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
Software update: The ORCA program system--Version 5.0 , author =. Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
-
[74]
The Journal of Chemical Physics , publisher =
The ORCA quantum chemistry program package , author =. The Journal of Chemical Physics , publisher =
-
[75]
Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
The ORCA program system , author =. Wiley Interdisciplinary Reviews: Computational Molecular Science , publisher =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.