Recognition: no theorem link
Path Dependence in Alchemical Calculations of Water Chemical Potential in Aqueous Electrolytes
Pith reviewed 2026-05-12 00:49 UTC · model grok-4.3
The pith
The order in which van der Waals and electrostatic interactions are activated during water insertion affects the computed chemical potential in aqueous salt solutions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
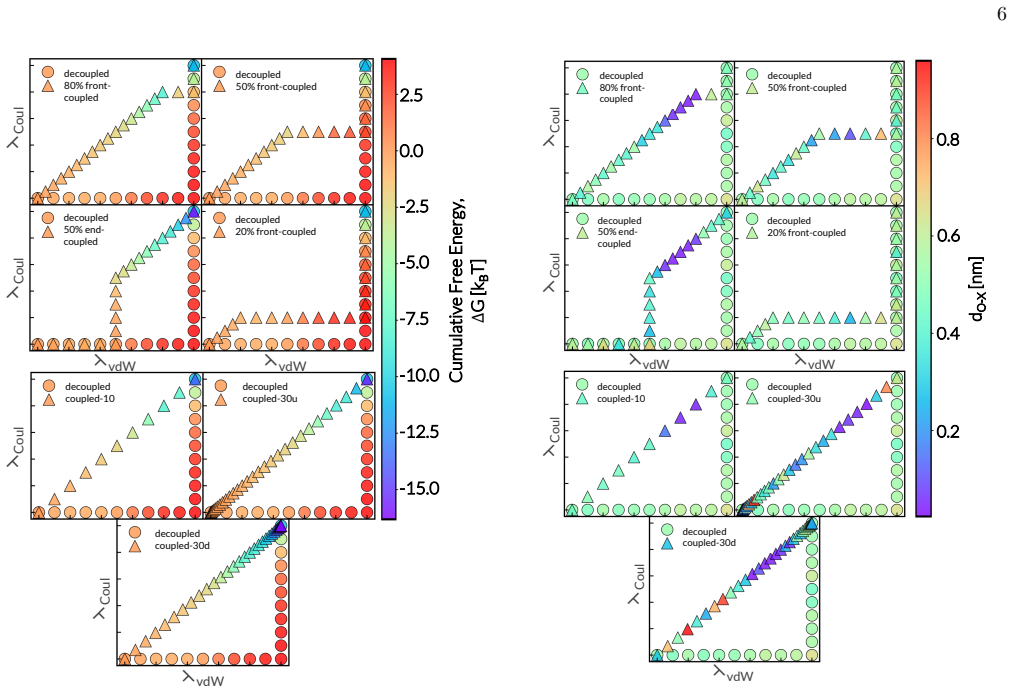
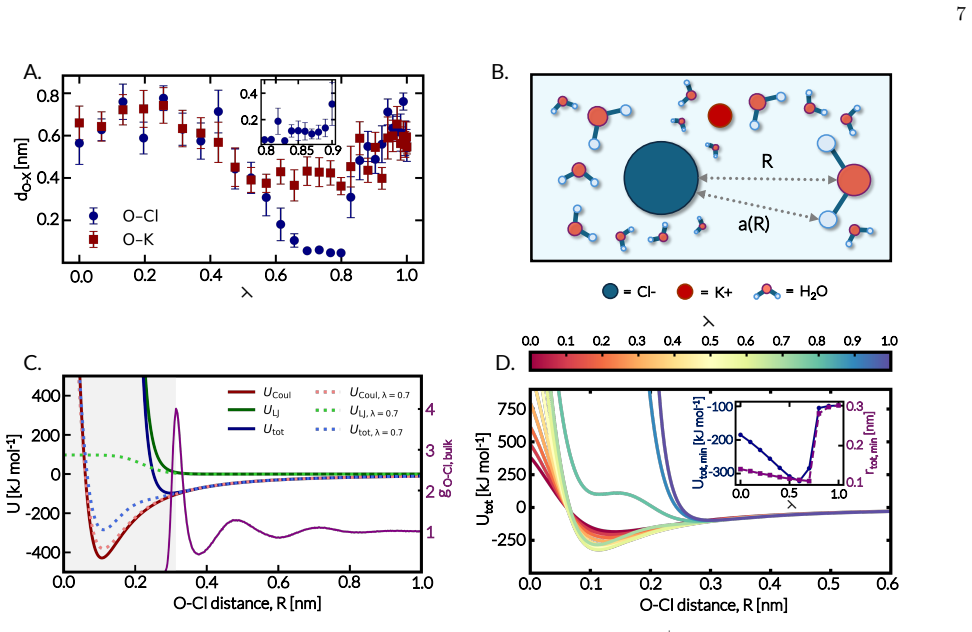
Although the insertion free energy of water should be independent of the alchemical pathway in principle, calculations in aqueous KCl show strong dependence on the order and extent of coupling van der Waals and Coulombic interactions. Concurrent or partially end-coupled protocols generate anomalous, implausible values that originate from intermediate states in which the inserted water molecule experiences strong electrostatic attraction to a chloride ion before adequate short-range repulsion is present. Protocols that activate short-range van der Waals interactions before electrostatics produce consistent and chemically reasonable estimates, demonstrating that practical alchemical protocols
What carries the argument
Staged particle-insertion alchemical protocols that vary the sequence of activating short-range van der Waals repulsions and long-range electrostatic attractions for an inserted water molecule.
If this is right
- Concurrent activation of van der Waals and electrostatic interactions can produce chemically implausible insertion free energies.
- Activating short-range van der Waals interactions before electrostatics yields consistent and plausible estimates.
- The chemical potential of water in ionic solutions is practically sensitive to the staging of interactions in alchemical protocols.
- Decoupling short-range repulsions from electrostatic attractions is required for reliable staged-insertion calculations in polar and charged environments.
Where Pith is reading between the lines
- The same pathway sensitivity is likely to appear when computing solvation free energies of other small molecules or ions in electrolyte solutions.
- Simulation protocols for charged systems should routinely compare results from at least two distinct staging orders to detect hidden artifacts.
- The findings suggest that similar decoupling strategies could improve convergence in alchemical transformations involving charged species or polar solvents.
- Free-energy calculations in complex media may require explicit validation against multiple insertion sequences before the results are treated as pathway-independent.
Load-bearing premise
The inconsistencies in the computed free energies are caused by the choice of alchemical pathway rather than by insufficient sampling, force-field errors, or finite-size effects.
What would settle it
Repeating the anomalous pathways with substantially longer simulations or enhanced sampling methods and finding that all pathways converge to the same free-energy value would show that the discrepancies are not inherent to the pathway design.
Figures




read the original abstract
Accurate calculation of free energies and their derivatives is central to assessing the thermodynamic stability of molecular and particulate systems across length scales. Yet such quantities can be difficult to compute reliably in strongly interacting systems, such as solutions of ionic species in polar solvents. One important example is the chemical potential of water in aqueous electrolytes, which can be estimated through staged particle insertion by gradually coupling an inserted molecule to its environment. Although the resulting insertion free energy should be independent of the alchemical pathway, the order and manner in which van der Waals and electrostatic interactions are activated can strongly affect convergence and, in some cases, yield inconsistent estimates. Here, we examine this issue by calculating water's chemical potential in aqueous KCl solutions using eight alchemical insertion pathways that differ in the extent and order of van der Waals and Coulombic coupling. We find that concurrently activating these interactions, particularly in fully coupled and partially end-coupled protocols, can produce chemically implausible insertion free energies. These anomalies arise from intermediate states in which the inserted water molecule develops strong electrostatic interactions with a chloride ion before sufficient short-range repulsion has been established. In contrast, pathways that activate short-range van der Waals interactions before electrostatics yield more consistent and chemically plausible estimates. These findings demonstrate that practical alchemical calculations in polar and ionic environments can be highly sensitive to pathway design, underscoring the importance of decoupling short-range and electrostatic interactions in staged insertion alchemical protocols.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript examines path dependence in alchemical free-energy calculations of the chemical potential of water in aqueous KCl solutions. By comparing eight staged-insertion pathways that differ in the order and extent of van der Waals versus electrostatic coupling, the authors report that protocols activating short-range repulsion before electrostatics produce consistent, chemically plausible insertion free energies, while concurrent or electrostatic-first pathways yield implausible values. These anomalies are attributed to intermediate lambda states in which the inserted water forms strong attractions to chloride ions before sufficient short-range repulsion is established.
Significance. If the central claim is substantiated with adequate convergence diagnostics, the work would be significant for the field of computational physical chemistry. It provides concrete evidence that alchemical pathway design can introduce artifacts in polar/ionic systems and offers practical guidance on decoupling short-range and electrostatic interactions. The systematic comparison of eight pathways is a methodological strength; however, the absence of error bars, overlap metrics, or sampling diagnostics currently limits the strength of the conclusions.
major comments (3)
- [Abstract] Abstract: The claim that electrostatic-first pathways produce 'chemically implausible insertion free energies' is central to the argument yet is presented without numerical values, uncertainties, or comparison to reference data (e.g., experimental or independent simulation results), making it impossible to judge the magnitude or statistical significance of the reported anomalies.
- [Results] Results (pathway comparison): The attribution of inconsistencies specifically to intermediate-state artifacts (strong water-Cl- electrostatics before repulsion) cannot be distinguished from inadequate sampling of high-energy barriers, because no overlap matrices, forward/reverse work histograms, lambda-window convergence tests, or autocorrelation times are reported; these diagnostics are load-bearing for the claim that the observed path dependence is physical rather than numerical.
- [Methods] Methods: Finite-size corrections, system-size dependence, and the precise lambda schedules (including number of windows and soft-core parameters) are not detailed; without these, it is unclear whether the reported differences arise from pathway ordering or from other simulation limitations such as periodic-boundary artifacts in the electrolyte.
minor comments (2)
- [Abstract] Abstract: The phrasing 'concurrently activating these interactions, particularly in fully coupled and partially end-coupled protocols' is ambiguous; explicit enumeration of which of the eight pathways fall into each category would improve clarity.
- [Results] The manuscript would benefit from a table summarizing the eight pathways (coupling order, lambda points, and resulting free energies with uncertainties) to allow direct comparison.
Simulated Author's Rebuttal
We thank the referee for their insightful comments on our manuscript. We address each of the major comments below and will revise the manuscript accordingly to enhance its clarity and completeness.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that electrostatic-first pathways produce 'chemically implausible insertion free energies' is central to the argument yet is presented without numerical values, uncertainties, or comparison to reference data (e.g., experimental or independent simulation results), making it impossible to judge the magnitude or statistical significance of the reported anomalies.
Authors: We agree that including numerical values and uncertainties would strengthen the abstract. In the revised version, we will incorporate specific examples of the insertion free energies obtained from the different pathways, including their statistical uncertainties. For context, we will also reference independent simulation results from the literature on water chemical potentials in electrolytes to illustrate the expected range and magnitude of the observed anomalies. revision: yes
-
Referee: [Results] Results (pathway comparison): The attribution of inconsistencies specifically to intermediate-state artifacts (strong water-Cl- electrostatics before repulsion) cannot be distinguished from inadequate sampling of high-energy barriers, because no overlap matrices, forward/reverse work histograms, lambda-window convergence tests, or autocorrelation times are reported; these diagnostics are load-bearing for the claim that the observed path dependence is physical rather than numerical.
Authors: We acknowledge that the lack of these diagnostics in the original submission makes it difficult to fully rule out sampling issues. However, the fact that several pathways yield mutually consistent results while others do not, and that the anomalous results correlate with the presence of unphysical intermediate states, supports our physical interpretation. To address this, we will include overlap matrices, forward/reverse histograms, and autocorrelation times in the revised manuscript to demonstrate adequate sampling in the well-behaved pathways. revision: yes
-
Referee: [Methods] Methods: Finite-size corrections, system-size dependence, and the precise lambda schedules (including number of windows and soft-core parameters) are not detailed; without these, it is unclear whether the reported differences arise from pathway ordering or from other simulation limitations such as periodic-boundary artifacts in the electrolyte.
Authors: We thank the referee for highlighting these omissions. The revised Methods section will provide full details on the lambda schedules, including the number of windows and soft-core parameters for each stage, as well as the system sizes employed and any finite-size corrections applied. We will clarify that the path dependence is observed consistently across our simulation setups, indicating it is not due to periodic-boundary artifacts in the electrolyte. revision: yes
Circularity Check
No circularity: empirical simulation results with no self-referential derivations
full rationale
The paper reports numerical outcomes from molecular dynamics simulations of eight alchemical insertion pathways for water chemical potential in KCl solutions. No equations or derivations are presented that reduce a claimed prediction or first-principles result to fitted parameters, self-definitions, or prior self-citations by construction. The central observation—that certain pathways produce implausible free energies due to intermediate-state artifacts—is an empirical finding from direct computation, not a closed mathematical loop. The work is self-contained against external benchmarks (standard alchemical theory and MD sampling diagnostics) and contains no load-bearing self-citation chains or ansatz smuggling.
Axiom & Free-Parameter Ledger
free parameters (1)
- alchemical coupling schedule
axioms (2)
- domain assumption The true insertion free energy is independent of the alchemical pathway provided sampling is sufficient and the end states are identical.
- domain assumption Standard force fields and periodic boundary conditions adequately represent aqueous KCl solutions for the purpose of detecting pathway-dependent artifacts.
Reference graph
Works this paper leans on
-
[1]
J. G. Kirkwood, Statistical mechanics of fluid mixtures, J. Chem. Phys.3, 300 (1935)
work page 1935
-
[2]
C. H. Bennett, Efficient estimation of free energy differ- ences from Monte Carlo data, J. Comput. Phys.22, 245 (1976)
work page 1976
-
[3]
S. Romano and K. Singer, Calculation of the entropy of liquid chlorine and bromine by computer simulation, Mol. Phys.37, 1765 (1979)
work page 1979
-
[4]
D. Frenkel and A. J. Ladd, New monte carlo method to compute the free energy of arbitrary solids. application to the fcc and hcp phases of hard spheres, J. Chem. Phys. 81, 3188 (1984)
work page 1984
-
[5]
Frenkel, Stability of the high-pressure body-centered- cubic phase of helium, Phys
D. Frenkel, Stability of the high-pressure body-centered- cubic phase of helium, Phys. Rev. Lett.56, 858 (1986)
work page 1986
-
[6]
B. A. Berg and T. Neuhaus, Multicanonical ensemble: A new approach to simulate first-order phase transitions, Phys. Rev. Lett.68, 9 (1992)
work page 1992
-
[7]
G. Orkoulas and A. Z. Panagiotopoulos, Free energy and phase equilibria for the restricted primitive model of ionic fluids from monte carlo simulations, J. Chem. Phys.101, 1452 (1994)
work page 1994
-
[8]
F. Wang and D. P. Landau, Efficient, multiple-range ran- dom walk algorithm to calculate the density of states, Phys. Rev. Lett.86, 2050 (2001)
work page 2050
-
[9]
A. Haji-Akbari, M. Engel, and S. C. Glotzer, Phase di- agram of hard tetrahedra, J. Chem. Phys.135, 194101 (2011)
work page 2011
-
[10]
J. Espinosa, C. Vega, and E. Sanz, The mold integration method for the calculation of the crystal-fluid interfacial free energy from simulations, J. Chem. Phys.141, 134709 (2014)
work page 2014
-
[11]
V. Thapar and F. A. Escobedo, Simultaneous estimation of free energies and rates using forward flux sampling and mean first passage times, J. Chem. Phys.143, 244113 (2015)
work page 2015
-
[12]
F. Sulantay Vargas, S. Hussain, and A. Haji-Akbari, Ro- bustness of classical nucleation theory to chemical hetero- geneity of crystal nucleating substrates, Cryst. Growth Des.26, 1943 (2026)
work page 1943
-
[13]
A. Pohorille, C. Jarzynski, and C. Chipot, Good practices in free-energy calculations, J. Phys. Chem. B114, 10235 (2010)
work page 2010
-
[14]
M. R. Shirts and D. L. Mobley, An introduction to best practices in free energy calculations, Methods Mol. Biol. 924, 271 (2013)
work page 2013
-
[15]
N. Hansen and W. F. Van Gunsteren, Practical Aspects of Free-Energy Calculations: A Review, J. Chem. Theory Comput.10, 2632 (2014)
work page 2014
-
[16]
P. V. Klimovich, M. R. Shirts, and D. L. Mobley, Guide- lines for the analysis of free energy calculations, J. Comput.-Aided Mol. Des.29, 397 (2015)
work page 2015
-
[17]
P. V. Klimovich and D. L. Mobley, A Python tool to set up relative free energy calculations in GROMACS, J. Comput.-Aided Mol. Des.29, 1007 (2015)
work page 2015
- [18]
-
[19]
S. Boresch, F. Tettinger, M. Leitgeb, and M. Karplus, Absolute binding free energies: a quantitative approach 10 for their calculation, J. Phys. Chem. B107, 9535 (2003)
work page 2003
-
[20]
J. C. Palmer, F. Martelli, Y. Liu, R. Car, A. Z. Pana- giotopoulos, and P. G. Debenedetti, Metastable liquid– liquid transition in a molecular model of water, Nature 510, 385 (2014)
work page 2014
-
[21]
the putative liquid-liquid transition is a liquid-solid transition in atomistic models of water
J. C. Palmer, A. Haji-Akbari, R. S. Singh, F. Martelli, R. Car, A. Z. Panagiotopoulos, and P. G. Debenedetti, Comment on “the putative liquid-liquid transition is a liquid-solid transition in atomistic models of water”[i and ii: J. chem. phys. 135, 134503 (2011); j. chem. phys. 138, 214504 (2013)], J. Chem. Phys.148, 137101 (2018)
work page 2011
-
[22]
S. H. Saravi and A. Z. Panagiotopoulos, Individual ion activity coefficients in aqueous electrolytes from explicit- water molecular dynamics simulations, J. Phys. Chem. B 125, 8511 (2021)
work page 2021
-
[23]
A. Kacirani, B. Uralcan, T. S. Domingues, and A. Haji- Akbari, Effect of Pressure on the Conformational Land- scape of HumanγD-Crystallin from Replica Exchange Molecular Dynamics Simulations, J. Phys. Chem. B128, 4931 (2024)
work page 2024
-
[24]
D. L. Beveridge and F. M. DiCapua, Free energy via molecular simulation: applications to chemical and biomolecular systems, Annu. Rev. Biophys. Biophys. Chem.18, 431 (1989)
work page 1989
-
[25]
J. W. Pitera and W. F. Van Gunsteren, A Comparison of Non-Bonded Scaling Approaches for Free Energy Cal- culations, Mol. Simul.28, 45 (2002)
work page 2002
-
[26]
R. W. Zwanzig, E. G. Rauh, and R. J. Thorn, High- Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases, J. Chem. Phys.22, 1420 (1954)
work page 1954
-
[27]
H. Resat and M. Mezei, Studies on free energy calcula- tions. I. Thermodynamic integration using a polynomial path, J. Chem. Phys.99, 6052 (1993)
work page 1993
- [28]
-
[29]
A. M. Ferrenberg and R. H. Swendsen, Optimized Monte Carlo data analysis, Phys. Rev. Lett.63, 1195 (1989)
work page 1989
-
[30]
M. R. Shirts and J. D. Chodera, Statistically optimal analysis of samples from multiple equilibrium states, J. Chem. Phys.129, 124105 (2008)
work page 2008
-
[31]
T. Koop, B. Luo, A. Tsias, and T. Peter, Water activ- ity as the determinant for homogeneous ice nucleation in aqueous solutions, Nature406, 611 (2000)
work page 2000
- [32]
-
[33]
J. Sauter and A. Grafm¨ uller, Predicting the chemical po- tential and osmotic pressure of polysaccharide solutions by molecular simulations, J. Chem. Theory Comput.12, 4375 (2016)
work page 2016
-
[34]
J. R. Espinosa, G. D. Soria, J. Ramirez, C. Valeriani, C. Vega, and E. Sanz, Role of salt, pressure, and water activity on homogeneous ice nucleation, J. Phys. Chem. Lett.8, 4486 (2017)
work page 2017
-
[35]
Widom, Some Topics in the Theory of Fluids, J
B. Widom, Some Topics in the Theory of Fluids, J. Chem. Phys.39, 2808 (1963)
work page 1963
-
[36]
D. Frenkel and B. Smit, Understanding molecular simu- lation: From algorithms to applications, Understanding Mol. Simul. 10.1063/1.881812 (1996)
-
[37]
S. G. Moore and D. R. Wheeler, Chemical potential per- turbation: A method to predict chemical potentials in pe- riodic molecular simulations, J. Chem. Phys.134, 114514 (2011)
work page 2011
- [38]
-
[39]
S. Qin and H.-X. Zhou, Fast method for computing chemical potentials and liquid–liquid phase equilibria of macromolecular solutions, J. Phys. Chem. B120, 8164 (2016)
work page 2016
-
[40]
K. Shing and K. Gubbins, The chemical potential in dense fluids and fluid mixtures via computer simulation, Mol. Phys.46, 1109 (1982)
work page 1982
-
[41]
G. C. Boulougouris, I. G. Economou, and D. N. Theodorou, On the calculation of the chemical poten- tial using the particle deletion scheme, Mol. Phys.96, 905 (1999)
work page 1999
-
[42]
R. Delgado-Buscalioni, G. De Fabritiis, and P. Coveney, Determination of the chemical potential using energy- biased sampling, J. Chem. Phys.123, 054105 (2005)
work page 2005
-
[43]
A. V. Neimark and A. Vishnyakov, A simulation method for the calculation of chemical potentials in small, in- homogeneous, and dense systems, J. Chem. Phys.122, 234108 (2005)
work page 2005
-
[44]
S. Qin and H.-X. Zhou, Fft-based method for modeling protein folding and binding under crowding: benchmark- ing on ellipsoidal and all-atom crowders, J. Chem. Theory Comput.9, 4633 (2013)
work page 2013
- [45]
-
[46]
D. R. Squire and W. G. Hoover, Monte carlo simulation of vacancies in rare-gas crystals, J. Chem. Phys.50, 701 (1969)
work page 1969
-
[47]
T. T. Pham and M. R. Shirts, Identifying low vari- ance pathways for free energy calculations of molecular transformations in solution phase, J. Chem. Phys.135, 10.1063/1.3607597 (2011)
-
[48]
D. K. Shenfeld, H. Xu, M. P. Eastwood, R. O. Dror, and D. E. Shaw, Minimizing thermodynamic length to select intermediate states for free-energy calculations and replica-exchange simulations, Phys. Rev. E80, 046705 (2009)
work page 2009
-
[49]
S. Decherchi and A. Cavalli, Optimal Transport for Free Energy Estimation, J. Phys. Chem. Lett.14, 1618 (2023)
work page 2023
-
[50]
H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura, and T. Head-Gordon, Develop- ment of an improved four-site water model for biomolec- ular simulations: TIP4P-Ew, J. Chem. Phys.120, 9665 (2004)
work page 2004
-
[51]
I. S. Joung and T. E. Cheatham III, Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations, J. Phys. Chem. B112, 9020 (2008)
work page 2008
-
[52]
M. J. Abraham, T. Murtola, R. Schulz, S. P´ all, J. C. Smith, B. Hess, and E. Lindahl, GROMACS: High per- formance molecular simulations through multi-level par- allelism from laptops to supercomputers, SoftwareX1-2, 19 (2015)
work page 2015
-
[53]
R. W. Hockney, The potential calculation and some ap- plications, Methods Comput. Phys.20, 135 (1970)
work page 1970
-
[54]
Nos´ e, A molecular dynamics method for simulations in the canonical ensemble, Mol
S. Nos´ e, A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys.52, 255 (1984)
work page 1984
-
[55]
W. G. Hoover, Canonical dynamics: Equilibrium phase- 11 space distributions, Phys. Rev. A31, 1695 (1985)
work page 1985
-
[56]
M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys.52, 7182 (1981)
work page 1981
-
[57]
M. Bernetti and G. Bussi, Pressure control using stochas- tic cell rescaling, J. Chem. Phys.153, 114107 (2020)
work page 2020
-
[58]
U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen, A smooth particle mesh Ewald method, J. Chem. Phys.103, 8577 (1995)
work page 1995
-
[59]
Hess, P-lincs: A parallel linear constraint solver for molecular simulation, J
B. Hess, P-lincs: A parallel linear constraint solver for molecular simulation, J. Chem. Theory Comput.4, 116 (2007)
work page 2007
-
[60]
A. Luzar and D. Chandler, Hydrogen-bond kinetics in liquid water, Nature379, 55 (1996)
work page 1996
-
[61]
D. van der Spoel, P. J. van Maaren, P. Larsson, and N. Tˆ ımneanu, Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media, J. Phys. Chem. B 110, 4393 (2006)
work page 2006
-
[62]
T. C. Beutler, A. E. Mark, R. C. van Schaik, P. R. Ger- ber, and W. F. van Gunsteren, Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations, Chem. Phys. Lett.222, 529 (1994)
work page 1994
-
[63]
H. Han, D. Li, L. Guo, Y. Yao, H. Yang, and D. Zeng, Isopiestic measurements of water activity for the NaCl– KCl–MgCl2–H2O systems at 323.15 K, J. Chem. Eng. Data60, 1139 (2015)
work page 2015
-
[64]
Chipot, Frontiers in free-energy calculations of biolog- ical systems, WIREs Comput
C. Chipot, Frontiers in free-energy calculations of biolog- ical systems, WIREs Comput. Mol. Sci.4, 71 (2014)
work page 2014
-
[65]
A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolin- tineanu, W. M. Brown, P. S. Crozier, P. J. In’t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen,et al., LAMMPS– a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comput. Phys. Commun.271, 108171 (2022)
work page 2022
-
[66]
D. A. Case, D. S. Cerutti, V. W. D. Cruzeiro, T. A. Dar- den, R. E. Duke, M. Ghazimirsaeed, G. M. Giambasu, T. J. Giese, A. W. Gotz, J. A. Harris,et al., Recent devel- opments in AMBER biomolecular simulations, J. Chem. Inf. Model.65, 7835 (2025)
work page 2025
-
[67]
W. R. Scott, P. H. H¨ unenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Hu- ber, P. Kr¨ uger, and W. F. Van Gunsteren, The GRO- MOS biomolecular simulation program package, J. Phys. Chem. A103, 3596 (1999)
work page 1999
-
[68]
A. A. Lashkov, I. V. Tolmachev, P. A. Eistrikh-Heller, and S. V. Rubinsky, PyFepRestr: Plugin to PyMOL Molecular Graphics System for Calculating the Free En- ergy of Ligand–Receptor Binding, Crystallogr. Rep.66, 861 (2021)
work page 2021
-
[69]
J. H. Moore, C. Margreitter, J. P. Janet, O. Engkvist, B. L. de Groot, and V. Gapsys, Automated relative bind- ing free energy calculations from SMILES to ∆∆G, Com- mun. Chem.6, 1 (2023)
work page 2023
-
[70]
M. T. Robo, R. L. Hayes, X. Ding, B. Pulawski, and J. Z. Vilseck, Fast free energy estimates fromλ-dynamics with bias-updated Gibbs sampling, Nat. Commun.14, 1 (2023)
work page 2023
-
[71]
S. W. Rick, Increasing the Efficiency of Free Energy Calculations Using Parallel Tempering and Histogram Reweighting, J. Chem. Theory Comput.2, 939 (2006)
work page 2006
- [72]
-
[73]
B. A. Shoemaker, T. S. Domingues, and A. Haji-Akbari, Ideal Conductor Model: An Analytical Finite-Size Cor- rection for Nonequilibrium Molecular Dynamics Simula- tions of Ion Transport through Nanoporous Membranes, J. Chem. Theory Comput.18, 7142 (2022)
work page 2022
-
[74]
B. A. Shoemaker and A. Haji-Akbari, Ideal conduc- tor/dielectric model (ICDM): A generalized technique to correct for finite-size effects in molecular simulations of hindered ion transport, J. Chem. Phys.160, 24116 (2024)
work page 2024
-
[75]
O. Khalifa, B. A. Shoemaker, and A. Haji-Akbari, Sec- ondary finite-size effects and multibarrier free energy landscapes in molecular simulations of hindered ion transport, J. Phys. Chem. B129, 11969 (2025)
work page 2025
-
[76]
M. Ghasemi and R. G. Larson, Future directions in phys- iochemical modeling of the thermodynamics of polyelec- trolyte coacervates, AIChE J.68, e17646 (2022)
work page 2022
-
[77]
Y. Luo, E. Harder, R. S. Faibish, and B. Roux, Computer simulations of water flux and salt permeability of the reverse osmosis ft-30 aromatic polyamide membrane, J. Membr. Sci.384, 1 (2011)
work page 2011
-
[78]
V. Kolev and V. Freger, Molecular dynamics investiga- tion of ion sorption and permeation in desalination mem- branes, J. Phys. Chem. B119, 14168 (2015)
work page 2015
- [79]
-
[80]
B. A. Shoemaker, O. Khalifa, and A. Haji-Akbari, Corre- lations in Charged Multipore Systems: Implications for Enhancing Selectivity and Permeability in Nanoporous Membranes, ACS Nano18, 1420 (2024)
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.