Recognition: 2 theorem links
· Lean TheoremMulti-Fidelity Computational Screening of High-Entropy MBenes for CO₂ Electroreduction
Pith reviewed 2026-05-12 02:11 UTC · model grok-4.3
The pith
A DFT-MLIP-AIMD screening pipeline identifies 45 thermodynamically stable high-entropy MBenes that support CO2 adsorption and reduction to CO.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
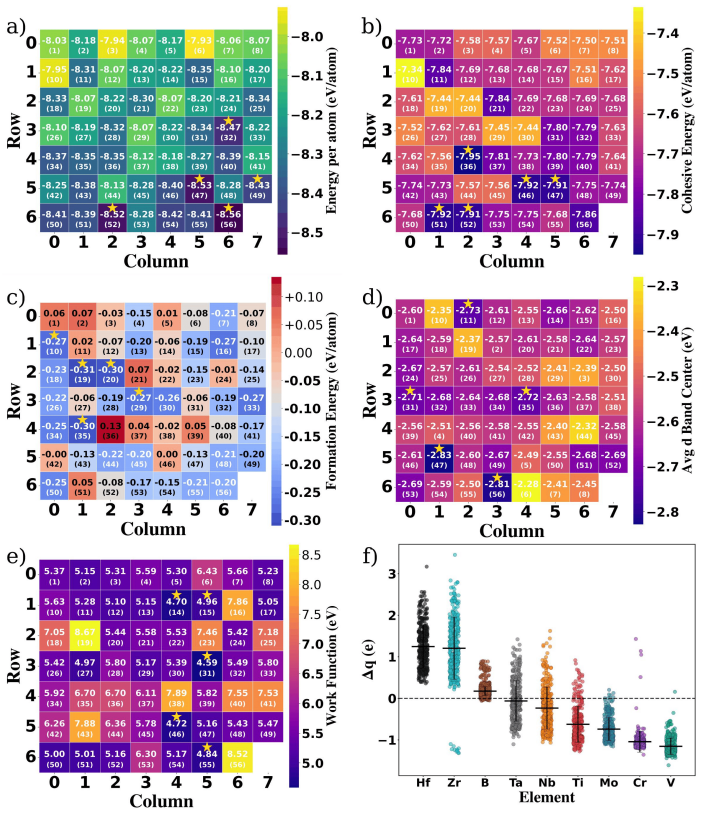
Through Monte Carlo special quasirandom structures, 56 quinary HE-MBene supercells are constructed from the {Ti, V, Cr, Mo, Nb, Ta, Zr, Hf} pool and relaxed with DFT (PBE+D3), of which 55 converge and 45 display negative formation energies. A MACE MLIP fine-tuned on this dataset reproduces adsorption and pristine energies with RMSEs of 3.49 and 3.0 meV/atom, permitting rapid identification of active metal sites via projected density-of-states matching between d-orbitals and CO2 molecular orbitals. The computational hydrogen electrode model is then applied to evaluate the rate-determining step of the CO2-to-CO pathway, while AIMD trajectories at 500 K over 2.5 ps assess short-time structural.
What carries the argument
MACE machine-learning interatomic potential fine-tuned on DFT data, used to predict CO2 adsorption energies and active sites across disordered high-entropy MBene surfaces.
If this is right
- 45 of the 56 compositions exhibit negative formation energies and are therefore thermodynamically stable.
- The MLIP enables efficient screening of CO2 binding on chemically disordered surfaces that would be computationally prohibitive with direct DFT.
- Active sites are localized on transition-metal atoms whose d-states overlap with CO2 frontier orbitals.
- The CHE model supplies a consistent ranking of the CO2-to-CO pathway across all candidates.
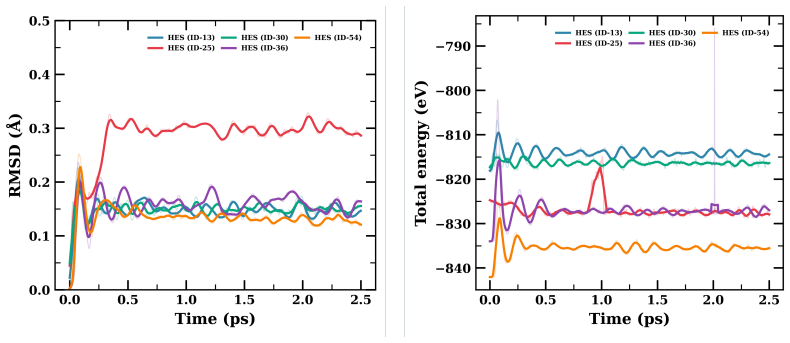
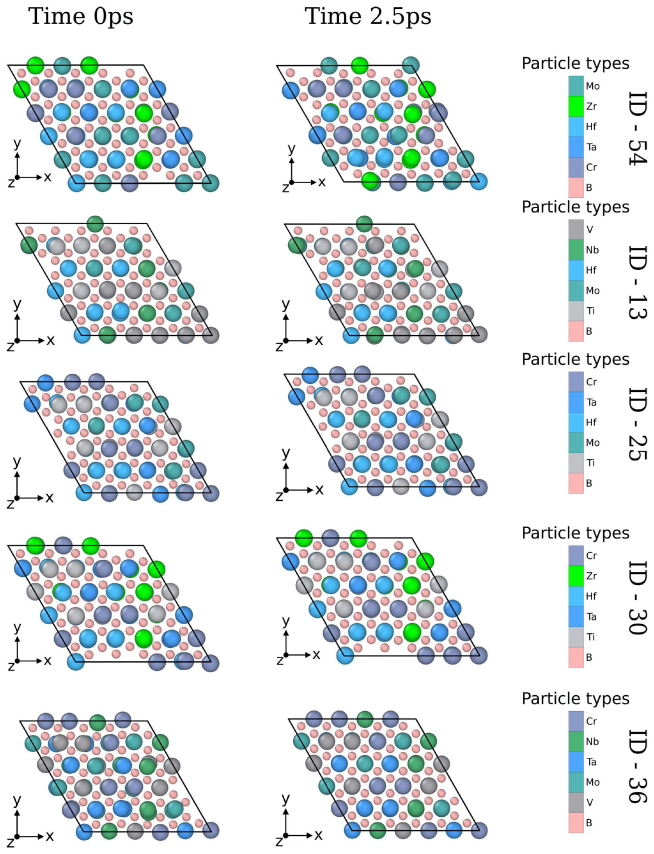
- Short AIMD runs establish that the relaxed structures maintain integrity at 500 K for at least 2.5 ps.
Where Pith is reading between the lines
- The same MLIP training strategy could be reused to screen adsorption of additional intermediates and thereby predict selectivity toward C2 or C3 products.
- Phonon calculations, flagged as future work, would be required to confirm that the 45 candidates remain dynamically stable at operating temperatures.
- The identified compositions provide concrete targets for experimental synthesis and electrochemical testing of HE-MBene electrodes.
- The multi-fidelity pipeline could be transferred to other high-entropy 2D families or to different catalytic reactions such as nitrogen reduction.
Load-bearing premise
The fine-tuned MACE MLIP reproduces DFT-level CO2 adsorption energies and correctly identifies active sites on disordered HE-MBene surfaces, and the computational hydrogen electrode model accurately ranks the rate-determining step for CO2-to-CO conversion.
What would settle it
Full DFT calculations on a random subset of the 45 stable candidates that yield adsorption energy errors well above the reported 3.5 meV/atom MLIP RMSE, or experimental synthesis of a top-ranked composition that shows no measurable CO production under CO2 electroreduction conditions.
Figures







read the original abstract
High-entropy MBenes (HE-MBenes) represent a promising, unexplored class of 2D materials for electrocatalysis. In this work, we present a systematic computational screening of 56 equiatomic quinary HE-MBene compositions from the {Ti, V, Cr, Mo, Nb, Ta, Zr, Hf} pool for CO$_2$ adsorption and electroreduction. Using the Monte Carlo Special Quasirandom Structure (MCSQS) algorithm, we generated disordered M$_1B_1$-type supercells and assessed structural stability via DFT (PBE+D3) in VASP. Of the 56 candidates, 55 passed relaxation, with 45 exhibiting negative formation energies, confirming thermodynamic stability. To efficiently screen CO$_2$ adsorption across disordered surfaces, we developed a machine-learning interatomic potential (MLIP) using the MACE architecture. Fine-tuned on our DFT dataset, the model achieved energy RMSEs of 3.49 and 3.0 meV/atom for adsorbed and pristine sets, respectively. Active sites were identified via PDOS analysis, matching metal d-orbital signatures with CO$_2$ molecular orbitals. The rate-determining step of the CO$_2$-to-CO pathway was evaluated using the computational hydrogen electrode (CHE) model. Short-time structural integrity was assessed via AIMD at 500 K over 2.5 ps; phonon-based stability remains a priority for future work. Our results establish an integrated DFT-MLIP-AIMD framework for the rational design of high-entropy 2D materials tailored for CO$_2$ conversion.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a multi-fidelity screening of 56 equiatomic quinary high-entropy MBenes (HE-MBenes) drawn from the {Ti, V, Cr, Mo, Nb, Ta, Zr, Hf} pool for CO₂ electroreduction. MCSQS-generated disordered M₁B₁ supercells are relaxed with DFT (PBE+D3), yielding 55 successful relaxations and 45 structures with negative formation energies. A MACE MLIP is fine-tuned on the DFT dataset (reported energy RMSE 3.49 meV/atom on adsorbed configurations) to enable high-throughput CO₂ adsorption screening; active sites are assigned via PDOS analysis and the CO₂-to-CO pathway is ranked with the computational hydrogen electrode (CHE) model. Short-time thermal stability is checked with AIMD at 500 K. The central claim is that the integrated DFT-MLIP-AIMD workflow establishes a rational-design framework for high-entropy 2D materials in CO₂ conversion.
Significance. If the MLIP predictions for adsorption energies and site rankings on disordered surfaces prove reliable, the work would supply a practical high-throughput protocol for exploring compositionally complex 2D electrocatalysts that are otherwise intractable with direct DFT. The combination of MCSQS disorder modeling, MLIP acceleration, and CHE pathway evaluation is a timely contribution to the high-entropy materials literature.
major comments (2)
- [Abstract and MLIP section] Abstract and MLIP training/validation section: the reported energy RMSE of 3.49 meV/atom on the adsorbed DFT set implies absolute energy errors of ~0.14–0.35 eV for typical 40–100-atom MCSQS supercells. Because CO₂ adsorption energy is a difference of three independent configurations, uncorrelated errors can reach ~0.5 eV—comparable to or larger than the binding-energy variations used to rank sites and compositions. No held-out validation set of MLIP versus DFT adsorption energies on disordered HE-MBene surfaces, nor any error-propagation analysis, is provided; this directly affects the reliability of the screening results that support the “rational design” claim.
- [Abstract and Results on structural stability] Abstract and stability results: while 55/56 structures are reported as successfully relaxed and 45 as having negative formation energies, the manuscript supplies no tabulated formation energies, no comparison against ordered or binary MBene baselines, and no error bars on the DFT energies. These omissions make it impossible to assess how close the “stable” candidates lie to the convex hull or whether the 45/56 fraction is robust to the chosen exchange-correlation functional.
minor comments (2)
- [Abstract] The abstract states that phonon-based stability “remains a priority for future work,” yet the AIMD runs are only 2.5 ps; a brief note on the expected phonon convergence criteria or supercell size used for the AIMD would clarify the scope of the current stability assessment.
- [Active-site identification] The PDOS-based active-site assignment is described only qualitatively; a short quantitative metric (e.g., overlap integral or projected DOS peak alignment) would strengthen the link between electronic structure and adsorption preference.
Simulated Author's Rebuttal
We thank the referee for their insightful and constructive comments. We address each major comment below and outline the revisions we will implement to enhance the manuscript.
read point-by-point responses
-
Referee: [Abstract and MLIP section] Abstract and MLIP training/validation section: the reported energy RMSE of 3.49 meV/atom on the adsorbed DFT set implies absolute energy errors of ~0.14–0.35 eV for typical 40–100-atom MCSQS supercells. Because CO₂ adsorption energy is a difference of three independent configurations, uncorrelated errors can reach ~0.5 eV—comparable to or larger than the binding-energy variations used to rank sites and compositions. No held-out validation set of MLIP versus DFT adsorption energies on disordered HE-MBene surfaces, nor any error-propagation analysis, is provided; this directly affects the reliability of the screening results that support the “rational design” claim.
Authors: We agree that validating the MLIP on adsorption energy differences for the disordered HE-MBene surfaces is crucial for supporting the screening results. The RMSE value is calculated on the total energies of the training configurations, which include both pristine and adsorbed structures. To address this, we will include in the revised manuscript a held-out validation set consisting of DFT-computed adsorption energies on selected disordered surfaces, along with a direct comparison to MLIP predictions. Additionally, we will provide an error propagation analysis to estimate the uncertainty in the CO₂ adsorption energies and site rankings. This will better substantiate the reliability of the MLIP-accelerated screening. revision: yes
-
Referee: [Abstract and Results on structural stability] Abstract and stability results: while 55/56 structures are reported as successfully relaxed and 45 as having negative formation energies, the manuscript supplies no tabulated formation energies, no comparison against ordered or binary MBene baselines, and no error bars on the DFT energies. These omissions make it impossible to assess how close the “stable” candidates lie to the convex hull or whether the 45/56 fraction is robust to the chosen exchange-correlation functional.
Authors: We appreciate this observation and will revise the manuscript to include a supplementary table with the formation energies for all relaxed structures. We will also add a discussion comparing our results to known stabilities of binary MBenes from the literature. Details on the DFT convergence criteria will be provided to address the robustness of the energies. However, constructing the full convex hull for these quinary systems is computationally prohibitive as it requires evaluating numerous competing phases, and we will clarify this limitation in the text. Similarly, while PBE+D3 is a standard choice for such screenings, a full functional sensitivity analysis across all 56 compositions is beyond the current scope but noted as future work. revision: partial
- Full convex hull construction to precisely determine the stability of the 45 candidates relative to all possible phases.
- Comprehensive testing of the results' dependence on the exchange-correlation functional for the entire set of 56 compositions.
Circularity Check
No significant circularity in the derivation chain
full rationale
The paper generates MCSQS supercells, computes formation energies and relaxations directly with DFT (PBE+D3), trains a MACE MLIP on that DFT dataset to accelerate adsorption-energy evaluation on additional disordered configurations, identifies sites with standard PDOS analysis, ranks the CO2-to-CO pathway with the external CHE model, and checks short-time stability with AIMD. None of these steps reduce the reported stabilities, adsorption rankings, or design conclusions to the inputs by construction; the MLIP functions as a surrogate with explicit RMSE metrics rather than a definitional tautology, and no self-citations or uniqueness theorems are invoked as load-bearing premises. The workflow remains self-contained against standard external benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption PBE+D3 functional provides sufficient accuracy for structural stability and formation energies of HE-MBenes
- domain assumption The computational hydrogen electrode model correctly identifies the rate-determining step for CO2-to-CO on these surfaces
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
To efficiently screen CO2 adsorption across disordered surfaces, we developed a machine-learning interatomic potential (MLIP) using the MACE architecture. Fine-tuned on our DFT dataset, the model achieved energy RMSEs of 3.49 and 3.0 meV/atom...
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The rate-determining step of the CO2-to-CO pathway was evaluated using the computational hydrogen electrode (CHE) model.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Huynh, Tai Thien and Dang, Nam Nguyen and Pham, Hau Quoc , title =. Small , year =
-
[2]
Di, Yaxin and Wang, Zhiqi and Wang, Guangqiu and Wang, Junjie , title =. Advanced Science , year =
-
[3]
Journal of Physical Chemistry Letters , year =
Bai, Xiuxia and Zhao, Zhonglong and Lu, Gang , title =. Journal of Physical Chemistry Letters , year =
-
[4]
Chen, Zhi Wen and Gariepy, Zachary and Chen, Lixin and Yao, Xue and Anand, Abu and Liu, Szu-Jia and Tetsassi Feugmo, Conrard Giresse and Tamblyn, Isaac and Singh, Chandra Veer , title =. ACS Catalysis , volume =. 2022 , doi =
work page 2022
-
[5]
Physical Chemistry Chemical Physics , year =
Lu, Xiaoqing and Hu, Yuying and Cao, Shoufu and Li, Jiao and Yang, Chunyu and Chen, Zengxuan and Wei, Shuxian and Liu, Siyuan and Wang, Zhaojie , title =. Physical Chemistry Chemical Physics , year =
-
[6]
Journal of Physical Chemistry Letters , year =
Xiao, Yi and Shen, Chen and Hadaeghi, Niloofar , title =. Journal of Physical Chemistry Letters , year =
-
[7]
Li, Mingxia and Zhang, Yaoyu and Gao, Dongyue and Li, Ying and Yu, Chao and Fang, Yi and Huang, Yang and Tang, Chengchun and Guo, Zhonglu , title =. ChemPhysChem , year =
-
[8]
Peng, Qiyuan and Chen, Anjie and Zhou, Peng and Sun, Yi and Meng, Lijuan and Fan, Li and Zhang, Xiuyun , title =. ACS Omega , year =
-
[9]
Advanced Functional Materials , year =
Zheng, Zhixuan and Cui, Zhijie and Peng, Wenchao and Liu, Jiapeng , title =. Advanced Functional Materials , year =
-
[10]
Zhu, Zeqi and Li, Zijun and Liu, Zihe and Gu, Chen and Zhang, Qingfeng and Wang, Longlu , title =. Small Methods , year =
-
[11]
Advanced Functional Materials , year =
Zhu, Jinliang and Yu, Xiaoling and Guo, Manchuan and Chen, Zhijie and Ni, Bing-Jie , title =. Advanced Functional Materials , year =
-
[12]
Pedersen, Jack K. and Batchelor, Thomas A. A. and Bagger, Alexander and Rossmeisl, Jan , title =. ACS Catalysis , year =
-
[13]
Rittiruam, Meena and Khamloet, Pisit and Ektarawong, Annop and Atthapak, Chayanon and Saelee, Tinnakorn and Khajondetchairit, Patcharaporn and Alling, Bj. Screening of. Applied Surface Science , year =
-
[14]
Machine-Learning-Accelerated Density Functional Theory Screening of
Rittiruam, Meena and Khamloet, Pisit and Tiwtusthada, Sirapat and Ektarawong, Annop and Saelee, Tinnakorn and Atthapak, Chayanon and Khajondetchairit, Patcharaporn and Alling, Bj. Machine-Learning-Accelerated Density Functional Theory Screening of. Applied Surface Science , year =
-
[15]
Wu, Qinglin and Pan, Meidie and Zhang, Shikai and Sun, Dongpeng and Yang, Yang and Chen, Dong and Weitz, David A. and Gao, Xiang , title =. Energies , year =
-
[16]
Journal of Physical Chemistry C , year =
Wang, Shuo and Li, Lei and Li, Jing and Yuan, Chengzong and Kang, Yao and Hui, Kwan San and Zhang, Jintao and Bin, Feng and Fan, Xi and Chen, Fuming and Hui, Kwun Nam , title =. Journal of Physical Chemistry C , year =
-
[17]
Journal of Colloid and Interface Science , year =
Yu, Jie and Zeng, Yabing and Chen, Junyao and Tan, Kai and Lin, Wei , title =. Journal of Colloid and Interface Science , year =
- [18]
-
[19]
The Journal of Physical Chemistry C , volume =
Lund, Colton and Xu, Jiayi and Liu, Cong , title =. The Journal of Physical Chemistry C , volume =. 2025 , doi =
work page 2025
-
[20]
Kresse, G. and Furthm\". Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set , journal =. 1996 , volume =
work page 1996
- [21]
-
[22]
Projector augmented-wave method , journal =
Bl\". Projector augmented-wave method , journal =. 1994 , volume =
work page 1994
-
[23]
Perdew, J. P. and Burke, K. and Ernzerhof, M. , title =. Phys. Rev. Lett. , year =
-
[24]
Grimme, S. and Ehrlich, S. and Goerigk, L. , title =. J. Comput. Chem. , year =
- [25]
-
[26]
Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode , journal =
N. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode , journal =. 2004 , volume =
work page 2004
-
[27]
Yuan, Hao and Li, Zhenyu and Yang, Jinlong , title =. J. Phys. Chem. C , year =
-
[28]
and Sharma, Peter Anand and Varma, Akash K
Gunda, Harini and Klebanoff, Leonard E. and Sharma, Peter Anand and Varma, Akash K. and Dolia, Varun and Jasuja, Kabeer and Stavila, Vitalie , title =. ACS Materials Lett. , year =
-
[29]
Khaledialidusti, Rasoul and Khazaei, Mohammad and Wang, Vei and Miao, Nanxi and Si, Chen and Wang, Jianfeng and Wang, Junjie , title =. J. Phys.: Condens. Matter , year =
-
[30]
Trends in Atmospheric Carbon Dioxide , year =
- [31]
-
[32]
van de Walle, A. and Tiwary, P. and de Jong, M. and Olmsted, D. L. and Asta, M. and Dick, A. and Shin, D. and Wang, Y. and Chen, L.-Q. and Liu, Z.-K. , title =. CALPHAD , year =
-
[33]
Sanchez, J. M. and Ducastelle, F. and Gratias, D. , title =. Physica A: Statistical Mechanics and its Applications , year =
-
[34]
Cowley, J. M. , title =. Physical Review , year =
- [35]
-
[36]
and Dzamba, Misko and Gao, Meng and Rizvi, Ammar and Zitnick, C
Barroso-Luque, Luis and Shuaibi, Muhammed and Fu, Xiang and Wood, Brandon M. and Dzamba, Misko and Gao, Meng and Rizvi, Ammar and Zitnick, C. Lawrence and Ulissi, Zachary W. , journal =. Open Materials 2024 (. 2024 , month =
work page 2024
-
[37]
Lu, X. and Zhang, Y. and Wang, J. and Li, Q. , title =. Phys. Chem. Chem. Phys. , year =
-
[38]
Li, H. and Chen, Z. and Zhao, P. and Sun, Y. , title =. J. Phys. Chem. C , year =
- [39]
-
[40]
A practical guide to machine learning interatomic potentials – Status and future , journal =. 2025 , issn =. doi:https://doi.org/10.1016/j.cossms.2025.101214 , url =
-
[41]
ACS Sustainable Chemistry & Engineering , volume=
Surfactant-Assisted Exfoliation of Tantalum Diboride (TaB2) for Electrochemical CO2 Reduction , author=. ACS Sustainable Chemistry & Engineering , volume=. 2025 , publisher=
work page 2025
-
[42]
Thomas A.A. Batchelor and Jack K. Pedersen and Simon H. Winther and Ivano E. Castelli and Karsten W. Jacobsen and Jan Rossmeisl , title =. Joule , volume =. 2019 , issn =. doi:https://doi.org/10.1016/j.joule.2018.12.015 , url =
-
[43]
Nellaiappan, Subramanian and Katiyar, Nirmal Kumar and Kumar, Ritesh and Parui, Arko and Malviya, Kirtiman Deo and Pradeep, K. G. and Singh, Abhishek K. and Sharma, Sudhanshu and Tiwary, Chandra Sekhar and Biswas, Krishanu , title =. ACS Catalysis , volume =. 2020 , doi =
work page 2020
-
[44]
Electronic factors determining the reactivity of metal surfaces , author=. Surface Science , volume=
-
[45]
Togo, Atsushi and Chaput, Laurent and Tadano, Terumasa and Tanaka, Isao , title =. J. Phys.: Condens. Matter , year =
-
[46]
ACS Applied Materials & Interfaces , volume=
Vacancy Rich TiB2 Nanosheets Promote Electrochemical Ammonia Synthesis , author=. ACS Applied Materials & Interfaces , volume=. 2024 , publisher=
work page 2024
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.