Recognition: 2 theorem links
· Lean TheoremAn ab initio approach to energy alignment and charge-state prediction of adsorbates on ultrathin insulators
Pith reviewed 2026-05-12 01:13 UTC · model grok-4.3
The pith
A modular ab initio scheme predicts energy-level alignment and charge states of adsorbates on oxide-metal substrates by adding separate GW, polarization, pinning, and dipole calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We present a theoretical approach to determine the energy-level alignment of adsorbates on oxide/metal substrates. Ionization potentials and electron affinities of the isolated adsorbates are obtained using GW calculations, electronic bandgap polarization is quantified through the quasiparticle renormalization caused by the substrate, Fermi-level pinning is evaluated within the integer charge transfer model, and work function shifts arising from Pauli pushback or from the adsorbate-metal dipole are determined from the local variations of the electrostatic potential. This computationally efficient framework paves the way for high-throughput screening of molecular qubits and organic electronic
What carries the argument
The additive decomposition of alignment into isolated GW ionization potentials and electron affinities, substrate-induced quasiparticle renormalization, integer charge transfer pinning, and electrostatic work-function shifts from potential variations.
If this is right
- Charge transfer and resulting unpaired spin states can be predicted for many adsorbates without running prohibitive full-system simulations.
- Bandgap narrowing and orbital re-ordering after charge transfer are captured through the separate polarization and pinning steps.
- High-throughput screening becomes practical for identifying adsorbates suitable for spin manipulation experiments.
- Each physical process contributing to alignment remains visible for interpretation and refinement.
Where Pith is reading between the lines
- The same modular breakdown could be tested on other hybrid systems such as organic layers on different metals to check transferability.
- Running the method alongside a few full simulations on small test cases would reveal the size of errors from neglected coupled relaxation.
- If the predictions hold, the approach could guide experimental selection of adsorbates to achieve targeted charge states for qubit prototypes.
- Incorporating limited structural relaxation into the isolated and interface steps might extend the method to cases where geometry changes matter.
Load-bearing premise
The separate calculations of isolated GW energies, substrate polarization, integer charge transfer pinning, and work-function shifts can be added together to give the correct alignment without missing coupled many-body or structural effects that appear only in a full adsorbate-oxide-metal simulation.
What would settle it
A direct comparison of the predicted alignment or charge state against scanning tunneling spectroscopy measurements on a concrete system such as a chosen molecule on MgO over Ag would falsify the method if the values disagree beyond the expected numerical accuracy.
Figures



read the original abstract
The rapid progress of electron spin resonance scanning tunneling microscopy experiments has enabled the manipulation of individual adsorbate spin states physisorbed on ultrathin oxide layers supported on metal substrates. Electron resonance requires unpaired spin density on the adsorbate, which can be achieved, for instance, through charge transfer from the supporting substrate. This requires the correct energy-level alignment between the energy levels of the adsorbate and the Fermi energy of the substrate. Experiments on molecules and single atoms adsorbed on metal-insulator systems have revealed complex phenomena, including electronic bandgap narrowing, charge transfer, Fermi-level pinning, and the re-ordering of adsorbate orbitals after charge transfer. Despite these advances, a predictive first-principles approach based on accurate methods such as quasiparticle GW, capable of capturing these effects without the prohibitive cost of full adsorbate/oxide/metal simulations, remains an open challenge. In this work, we present a theoretical approach to determine the energy-level alignment of adsorbates on oxide/metal substrates. Our method transparently exposes all physical processes and strikes a balance between computational cost and accuracy. Ionization potentials and electron affinities of the isolated adsorbates are obtained using GW calculations, electronic bandgap polarization is quantified through the quasiparticle renormalization caused by the substrate, Fermi-level pinning is evaluated within the integer charge transfer model, and work function shifts arising from Pauli pushback or from the adsorbate-metal dipole are determined from the local variations of the electrostatic potential. This computationally efficient framework paves the way for highthroughput screening of molecular qubits and organic electronic interfaces.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes a decomposed ab initio workflow to predict energy-level alignment and charge states of adsorbates on ultrathin oxide/metal substrates. It combines (i) GW ionization potentials and electron affinities computed for the isolated adsorbate, (ii) quasiparticle renormalization of the adsorbate gap due to substrate polarization, (iii) Fermi-level pinning via the integer charge transfer model, and (iv) work-function shifts extracted from local electrostatic-potential variations, with the goal of reproducing experimental phenomena such as bandgap narrowing and orbital reordering at lower cost than full supercell simulations.
Significance. If the additivity of these four contributions can be shown to hold with controlled error, the framework would supply a transparent, computationally tractable route to high-throughput screening of molecular qubits and organic interfaces on metal-insulator systems, addressing a recognized gap between full GW supercell calculations and simpler model Hamiltonians.
major comments (1)
- [Abstract (workflow description)] The central claim that the four-term decomposition reproduces full-system alignment rests on the untested assumption that cross terms (adsorbate-induced changes in oxide polarization, structural relaxation feedback from charge transfer, and fractional-charge or dynamical-screening corrections) remain negligible. No quantitative error bound obtained from a reference full-supercell GW calculation on the same geometry is provided, leaving the accuracy of the additivity hypothesis unverified.
minor comments (1)
- [Abstract] The abstract states that the method 'transparently exposes all physical processes,' yet the precise definition of the integer-charge-transfer pinning energy and the procedure for extracting the local electrostatic shift are not given explicitly; a short methods subsection with the relevant formulas would improve clarity.
Simulated Author's Rebuttal
We thank the referee for their thorough review and positive assessment of the significance of our work. We address the major comment in detail below and have revised the manuscript accordingly.
read point-by-point responses
-
Referee: [Abstract (workflow description)] The central claim that the four-term decomposition reproduces full-system alignment rests on the untested assumption that cross terms (adsorbate-induced changes in oxide polarization, structural relaxation feedback from charge transfer, and fractional-charge or dynamical-screening corrections) remain negligible. No quantitative error bound obtained from a reference full-supercell GW calculation on the same geometry is provided, leaving the accuracy of the additivity hypothesis unverified.
Authors: We agree with the referee that the additivity of the four contributions is a key assumption in our approach, and that a quantitative validation against full-supercell GW calculations would be desirable. Unfortunately, the computational expense of performing GW calculations on the complete adsorbate/oxide/metal supercell precludes such a direct comparison at present. To address this concern, we have added a new subsection in the revised manuscript discussing the potential cross terms and providing bounds on their magnitude based on separate calculations and prior literature. Additionally, we have included more detailed comparisons with experimental results to support the overall accuracy of the method. revision: yes
Circularity Check
No circularity: workflow decomposes into independent external calculations without reduction to fitted inputs or self-definitions
full rationale
The presented method computes isolated-adsorbate GW ionization potentials/electron affinities, substrate-induced quasiparticle renormalization of the gap, integer-charge-transfer pinning to the Fermi level, and electrostatic work-function shifts from local potential variations, then adds the terms. None of these quantities is defined in terms of the final alignment, fitted to the target data, or derived by construction from the sum; each step invokes an established external technique (GW, ICT model) whose assumptions are stated rather than internally enforced. No equations, self-citations, or uniqueness theorems are shown that would collapse the result back onto its inputs. The derivation therefore remains self-contained against external benchmarks and does not exhibit any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption GW calculations on isolated adsorbates yield accurate ionization potentials and electron affinities
- domain assumption The integer charge transfer model correctly captures Fermi-level pinning at the interface
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Ionization potentials and electron affinities of the isolated adsorbates are obtained using GW calculations, electronic bandgap polarization is quantified through the quasiparticle renormalization caused by the substrate, Fermi-level pinning is evaluated within the integer charge transfer model, and work function shifts ... are determined from the local variations of the electrostatic potential.
-
IndisputableMonolith/Foundation/BranchSelection.leanbranch_selection contradicts?
contradictsCONTRADICTS: the theorem conflicts with this paper passage, or marks a claim that would need revision before publication.
Our method transparently exposes all physical processes and strikes a balance between computational cost and accuracy.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
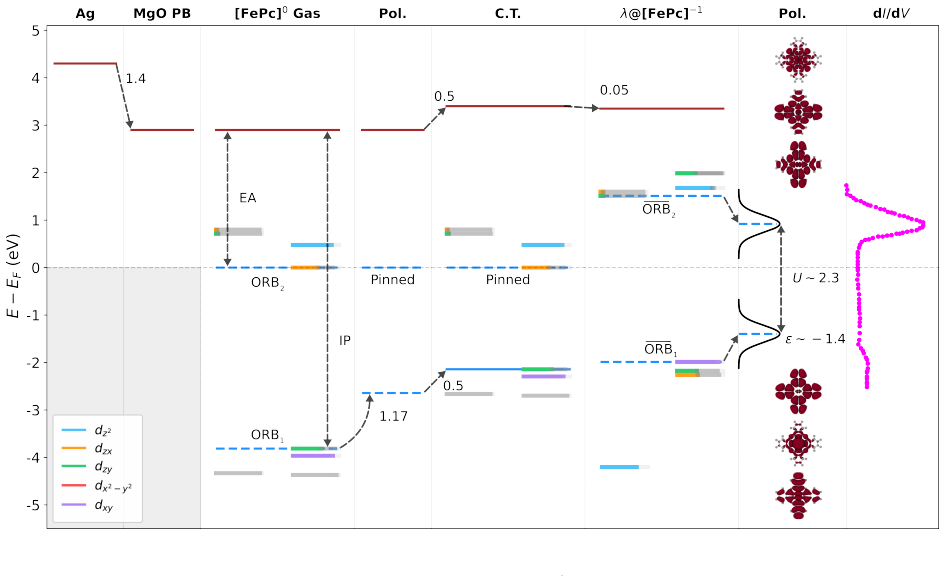
Fermi Level Pinning On the other hand, when a renormalized LUMO or HOMO approaches the Fermi level, charge transfer occurs, inducing an image charge in the metallic substrate [28, 30, 32]. The changes in the electric potential produced by this dipole lead to a shift of the vacuum level, ∆ϕ[13, 62, 63]. When the LUMO (HOMO) level becomes pinned, ∆ϕincrease...
-
[2]
Local Potential Variations and Impact on Energy Level Alignment The measured work function shift, ∆ϕ, depends strongly on the experimental probe. In localmeasurements such as STM/STS, ∆ϕincreases rapidly as the tip approaches the adsor- bate, reflecting the strong sensitivity to the local electrostatic environment [65]. In contrast, non-localtechniques su...
-
[3]
SUMO/SOMO Orbital splitting After charge transfer the molecular orbitals are re-organized. The energy level that gets pinned to the Fermi energy, splits into the Singly Occupied (Unoccupied) Molecular Orbital SOMO (SUMO) [13, 15, 66]. The energy difference between these orbitals is the screened electronic bandgap probed by local measurements such as scann...
-
[4]
Benzene on NaCl/Cu We start with the small closed shell organic molecule, benzene (C 6H6). To determine the energy alignment of benzene on NaCl/Cu, we begin with the determination of the work function for NaCl/Cu(001). The work function of Cu(001) sets the vacuum level to 5.0 eV, whilst 2 ML of NaCl decreases the vacuum level by ∆ϕ PB = 1.2 eV, in good ag...
-
[5]
FePc on MgO/Ag Neutral Iron(II)phthalocyanine (FePc) is a 3d 6,S= 1 system [70, 86]. We align the vacuum level using the work function of the Ag(001) interface,ϕ= 4.3 eV, and account for two monolayers of MgO, which introduce a Pauli pushback of ∆ϕ PB = 1.4 eV. The 3d transition metal core of FePc leads to a complicated orbital structure which could under...
-
[6]
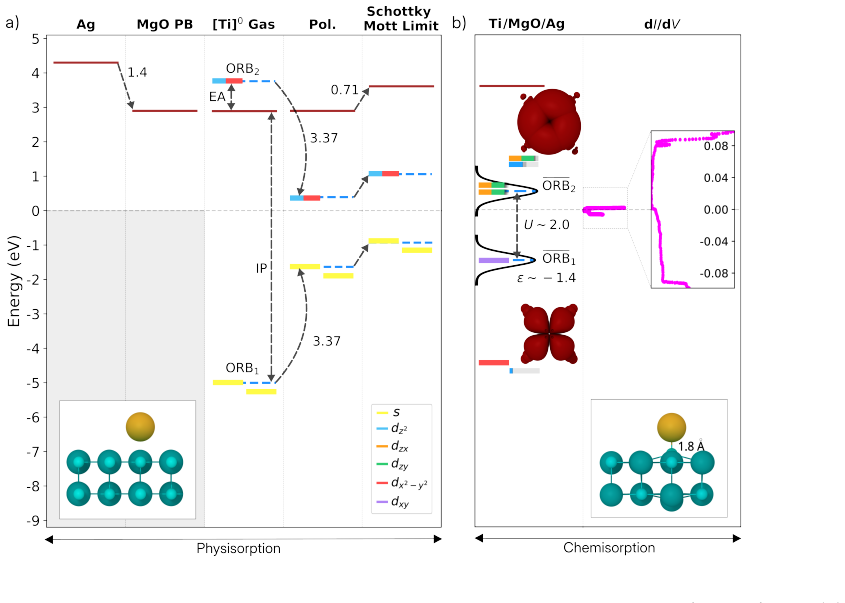
Ti atoms on MgO/Ag Lastly, we turn to single-atom adsorbates. Individual transition metal and lanthanide atoms have been studied using ESR-STM with the goal of building atomic-scale quantum bits. Among the 3dtransition metal series, particularly titanium (Ti) atoms on 2 and 3 ML of MgO have attracted interest due to their ability to formS= 1/2 prototypica...
-
[7]
Notes on GW parameters We have performed converged calculations within the framework of the GW COHSEX approximation for the isolated molecules. For non–transition-metal adsorbates such as ben- zene, pentacene, PTCDA, and TCNE, convergence was achieved using a cutoff for the ex- change–correlation potential matrix elements (VXCRLvcs) of 300000 RL, a cutoff...
-
[8]
Comparison of GW calculations and∆SCF calculations Fundamental electronic gaps of atoms and molecules can also be calculated by the so- called ∆SCF method, which uses the total energies of SCF calculations with different charge states to estimate the energy of removing (adding) one electron, i.e. the EA and IP. Figure 24 S2 shows the comparison of the ∆SC...
-
[9]
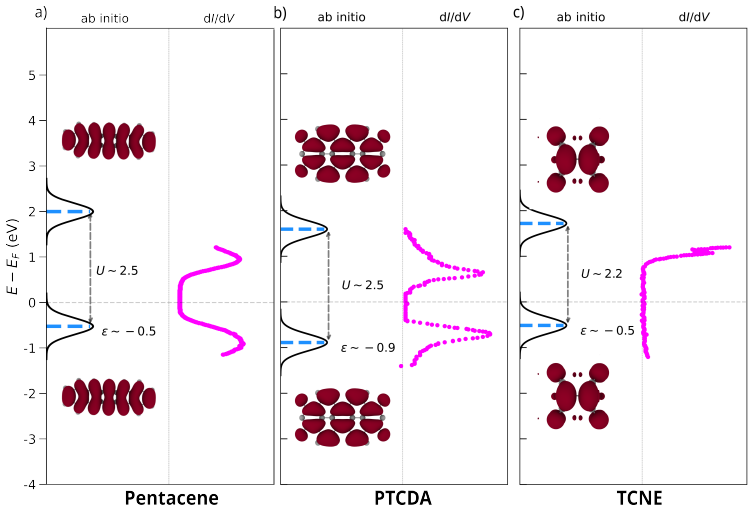
Detailed alignment plots for pentacene For pentacene deposited on MgO/Ag, we align the vacuum level to the work function of the interface Ag(001)ϕ= 4.3 eV and two monolayers of MgO with a Pauli pushback of ∆ϕPB = 1.4 eV. The alignment of the HOMO and LUMO levels (where the IP= 6.61 eV and EA= 1.35 eV), produces a LUMO energy level located at∼1 eV above th...
-
[10]
Detailed alignment plots for PTCDA Pristine PTCDA (C24H8O6) is a closed shell system with IP= 8.65 eV and EA= 3.20 eV and a electronic bandgap of 5.05 eV [36]. The alignment with respect to the vacuum level of the NaCl/Ag(111), located 3.85 eV above the Fermi energy–after the PB effect of ∆ϕ PB = 0.55–, does not initially indicate charge transfer upon ads...
-
[11]
The LUMO of TCNE immediately gets pinned to the Fermi 31 FIG
Detailed alignment plots for TCNE TCNE (C2(CN)4) is a closed-shell molecule characterized by a large ionization potential (IP= 12.33 eV) and a substantial electron affinity (EA= 3.51 eV), reflecting its strong electron-acceptor character. The LUMO of TCNE immediately gets pinned to the Fermi 31 FIG. S7.Energy level alignment of PTCDA on Ag/NaCl.The schema...
-
[12]
When aligned with Ag/MgO, neither level lies close to the Fermi energy
Detailed alignment plots for Ti Gas-phase Ti exhibits an ionization potential and electron affinity of 7.89 eV and -0.86 eV, respectively. When aligned with Ag/MgO, neither level lies close to the Fermi energy. 32 FIG. S8.Energy level alignment of TCNE on MgO/Ag.The schematic from left to right correspond to all the effects such as Pauli pushback (∆ϕ PB),...
-
[13]
A. J. Heinrich, W. D. Oliver, L. M. K. Vandersypen, A. Ardavan, R. Sessoli, D. Loss, A. B. Jayich, J. Fernandez-Rossier, A. Laucht, and A. Morello, Quantum-coherent nanoscience, Nature Nanotechnology16, 1318 (2021). 33
work page 2021
-
[14]
Y. Chen, Y. Bae, and A. J. Heinrich, Harnessing the quantum behavior of spins on surfaces, Advanced Materials35, 10.1002/adma.202107534 (2022)
-
[15]
A. Maiellaro, H. Aubin, A. Mesaros, and P. Simon, Local dynamics and detection of topology in spin-1 chains, Phys. Rev. B110, L220410 (2024)
work page 2024
-
[16]
C. Wolf, A. J. Heinrich, and S.-h. Phark, On-surface atomic scale qubit platform, ACS Nano 18, 28469–28479 (2024)
work page 2024
-
[17]
S. Baumann, W. Paul, T. Choi, C. P. Lutz, A. Ardavan, and A. J. Heinrich, Electron para- magnetic resonance of individual atoms on a surface, Science350, 417–420 (2015)
work page 2015
-
[18]
S. M¨ ullegger, E. Rauls, U. Gerstmann, S. Tebi, G. Serrano, S. Wiespointner-Baumgarthuber, W. G. Schmidt, and R. Koch, Mechanism for nuclear and electron spin excitation by radio frequency current, Phys. Rev. B92, 220418 (2015)
work page 2015
-
[19]
K. Yang, W. Paul, S.-H. Phark, P. Willke, Y. Bae, T. Choi, T. Esat, A. Ardavan, A. J. Heinrich, and C. P. Lutz, Coherent spin manipulation of individual atoms on a surface, Science366, 509–512 (2019)
work page 2019
-
[20]
F. D. Natterer, K. Yang, W. Paul, P. Willke, T. Choi, T. Greber, A. J. Heinrich, and C. P. Lutz, Reading and writing single-atom magnets, Nature543, 226–228 (2017)
work page 2017
-
[21]
D.-J. Choi, S.-h. Phark, A. J. Heinrich, and N. Lorente, Electron spin resonance with scan- ning tunneling microscopy: a tool for an on-surface quantum platform of identical qubits, Nanoscale Advances7, 4551–4558 (2025)
work page 2025
-
[22]
Z. Guo, J. Zhang, and Y. Chen, Electron spin resonance scanning tunneling microscopy beyond a single spin, Newton , 100413 (2026)
work page 2026
-
[23]
W. Liu, A. Tkatchenko, and M. Scheffler, Modeling adsorption and reactions of organic molecules at metal surfaces, Accounts of Chemical Research47, 3369–3377 (2014)
work page 2014
-
[24]
R. Kawaguchi, K. Hashimoto, T. Kakudate, K. Katoh, M. Yamashita, and T. Komeda, Spa- tially resolving electron spin resonance ofπ-radical in single-molecule magnet, Nano Letters 23, 213 (2023), pMID: 36585948, https://doi.org/10.1021/acs.nanolett.2c04049
-
[25]
M. Hollerer, D. L¨ uftner, P. Hurdax, T. Ules, S. Soubatch, F. S. Tautz, G. Koller, P. Puschnig, M. Sterrer, and M. G. Ramsey, Charge transfer and orbital level alignment at inor- ganic/organic interfaces: The role of dielectric interlayers, ACS Nano11, 6252 (2017), pMID: 28541656, https://doi.org/10.1021/acsnano.7b02449
- [26]
- [27]
- [28]
- [29]
-
[30]
L. Colazzo, C. Urdaniz, Y. Jung, L. Fang, S.-h. Phark, C. Wolf, W.-D. Schneider, A. Heinrich, and W.-J. Jang, Engineering spin interaction channels of fepc on au(111), Nano Letters25, 1883–1889 (2025)
work page 2025
-
[31]
S. Kovarik, R. Schlitz, A. Vishwakarma, D. Ruckert, P. Gambardella, and S. Stepanow, Spin torque–driven electron paramagnetic resonance of a single spin in a pentacene molecule, Science384, 1368 (2024), https://www.science.org/doi/pdf/10.1126/science.adh4753
- [32]
- [33]
-
[34]
K. Yang, Y. Bae, W. Paul, F. D. Natterer, P. Willke, J. L. Lado, A. Ferr´ on, T. Choi, J. Fern´ andez-Rossier, A. J. Heinrich, and C. P. Lutz, Engineering the eigenstates of coupled spin-1/2 atoms on a surface, Phys. Rev. Lett.119, 227206 (2017)
work page 2017
-
[35]
P. Willke, W. Paul, F. D. Natterer, K. Yang, Y. Bae, T. Choi, J. Fern´ andez-Rossier, A. J. Heinrich, and C. P. Lutz, Probing quantum coherence in single-atom electron spin resonance, Science Advances4, 10.1126/sciadv.aaq1543 (2018)
-
[36]
T. S. Seifert, S. Kovarik, D. M. Juraschek, N. A. Spaldin, P. Gambardella, and S. Stepanow, Longitudinal and transverse electron paramagnetic resonance in a scanning tunneling micro- scope, Science Advances6, 10.1126/sciadv.abc5511 (2020). 35
-
[37]
G. Czap, K. Noh, J. Velasco, R. M. Macfarlane, H. Brune, and C. P. Lutz, Direct electrical access to the spin manifolds of individual lanthanide atoms, ACS Nano19, 3705–3713 (2025)
work page 2025
-
[38]
S. Reale, J. Hwang, J. Oh, H. Brune, A. J. Heinrich, F. Donati, and Y. Bae, Electrically driven spin resonance of 4f electrons in a single atom on a surface, Nature Communications 15, 10.1038/s41467-024-49447-y (2024)
- [39]
-
[40]
M. T. Greiner, M. G. Helander, W.-M. Tang, Z.-B. Wang, J. Qiu, and Z.-H. Lu, Universal energy-level alignment of molecules on metal oxides, Nature Materials11, 76–81 (2011)
work page 2011
-
[41]
M. T. Greiner and Z.-H. Lu, Thin-film metal oxides in organic semiconductor devices: their electronic structures, work functions and interfaces, NPG Asia Materials5, e55–e55 (2013)
work page 2013
-
[42]
L. Ley, Y. Smets, C. I. Pakes, and J. Ristein, Calculating the universal energy-level alignment of organic molecules on metal oxides, Advanced Functional Materials23, 794–805 (2012)
work page 2012
-
[43]
P. Li and Z.-H. Lu, Interface engineering in organic electronics: Energy-level alignment and charge transport, Small Science1, 10.1002/smsc.202000015 (2020)
-
[44]
L. Chai, R. T. White, M. T. Greiner, and Z. H. Lu, Experimental demonstration of the universal energy level alignment rule at oxide/organic semiconductor interfaces, Phys. Rev. B89, 035202 (2014)
work page 2014
-
[45]
J. B. Neaton, M. S. Hybertsen, and S. G. Louie, Renormalization of molecular electronic levels at metal-molecule interfaces, Phys. Rev. Lett.97, 216405 (2006)
work page 2006
-
[46]
J. M. Garcia-Lastraet al., Polarization-induced renormalization of molecular levels at metal- lic and semiconducting surfaces, Phys. Rev. B80, 245427 (2009)
work page 2009
-
[47]
A. F¨ orster and F. Bruneval, Why does the gw approximation give accurate quasiparticle energies? the cancellation of vertex corrections quantified, The Journal of Physical Chemistry Letters15, 12526 (2024), pMID: 39670751, https://doi.org/10.1021/acs.jpclett.4c03126
-
[48]
Y. Kanget al., Ab initio calculation of ionization potential and electron affinity in solid-state organic semiconductors, Physical Review B93, 035131 (2016)
work page 2016
-
[49]
F. H¨ user, T. Olsen, and K. S. Thygesen, Quasiparticle gw calculations for solids, molecules, and two-dimensional materials, Phys. Rev. B87, 235132 (2013). 36
work page 2013
-
[50]
X. Blaseet al., First-principles gw calculations for fullerenes, porphyrins, phtalocyanine, and other molecules of interest for organic photovoltaic applications, Physical Review B83, 115103 (2011)
work page 2011
-
[51]
C. Rostgaard, K. W. Jacobsen, and K. S. Thygesen, Fully self-consistent gw calculations for molecules, Phys. Rev. B81, 085103 (2010)
work page 2010
-
[52]
F. Bruneval and M. A. L. Marques, Benchmarking the starting points of the gw approxima- tion for molecules, Journal of Chemical Theory and Computation9, 324–329 (2012)
work page 2012
- [53]
-
[54]
Mansouriet al., Gw approximation for open-shell molecules: a first-principles study, New J
M. Mansouriet al., Gw approximation for open-shell molecules: a first-principles study, New J. Phys.23, 093027 (2021)
work page 2021
- [55]
-
[56]
J. W. Knight, X. Wang, L. Gallandi, O. Dolgounitcheva, X. Ren, J. V. Ortiz, P. Rinke, T. K¨ orzd¨ orfer, and N. Marom, Accurate ionization potentials and electron affinities of accep- tor molecules iii: A benchmark of gw methods, Journal of Chemical Theory and Computation 12, 615–626 (2016)
work page 2016
-
[57]
S. K¨ orbel, P. Boulanger, I. Duchemin, X. Blase, M. A. L. Marques, and S. Botti, Benchmark many-body gw and bethe–salpeter calculations for small transition metal molecules, Journal of Chemical Theory and Computation10, 3934–3943 (2014)
work page 2014
-
[58]
N. D. Lang and W. Kohn, Theory of metal surfaces: Charge density and surface energy, Phys. Rev. B1, 4555 (1970)
work page 1970
-
[59]
H. L¨ uth, Adsorption on solid surfaces, inSolid Surfaces, Interfaces and Thin Films(Springer Berlin Heidelberg, 2001) p. 499–542
work page 2001
-
[60]
U. Martinez, L. Giordano, and G. Pacchioni, Tuning the work function of ultrathin ox- ide films on metals by adsorption of alkali atoms, The Journal of Chemical Physics128, 10.1063/1.2905218 (2008)
- [61]
- [62]
-
[63]
M. Bieletzki, T. Hynninen, T. M. Soini, M. Pivetta, C. R. Henry, A. S. Foster, F. Esch, C. Barth, and U. Heiz, Topography and work function measurements of thin mgo(001) films on ag(001) by nc-afm and kpfm, Phys. Chem. Chem. Phys.12, 3203 (2010)
work page 2010
-
[64]
S. Baumann, I. G. Rau, S. Loth, C. P. Lutz, and A. J. Heinrich, Measuring the three- dimensional structure of ultrathin insulating films at the atomic scale, ACS Nano8, 1739 (2014)
work page 2014
-
[65]
C. Wolf, F. Delgado, J. Reina, and N. Lorente, Efficient ab initio multiplet calculations for magnetic adatoms on mgo, The Journal of Physical Chemistry A124, 2318 (2020)
work page 2020
-
[66]
H. Wang, M. Oehzelt, S. Winkler, R. Ovsyannikov, N. Koch, and P. Amsalem, Electronic properties and degradation upon vuv irradiation of sodium chloride on ag(111) studied by photoelectron spectroscopy, Electronic Structure3, 034008 (2021)
work page 2021
-
[67]
M. Robledo, G. Pacchioni, F. Mart´ ın, M. Alcam´ ı, and S. D´ ıaz-Tendero, Adsorption of ben- zene on cu(100) and on cu(100) covered with an ultrathin nacl film: Molecule–substrate interaction and decoupling, The Journal of Physical Chemistry C119, 4062 (2015), https://doi.org/10.1021/jp5106604
-
[68]
N. Nilius, Properties of oxide thin films and their adsorption behavior studied by scanning tunneling microscopy and conductance spectroscopy, Surface Science Reports64, 595–659 (2009)
work page 2009
-
[69]
S. B. Cho, K.-H. Yun, D. S. Yoo, K. Ahn, and Y.-C. Chung, Work function tuning of an ultrathin mgo film on an ag substrate by generating oxygen impurities at the interface, Thin Solid Films544, 541 (2013), the 6th International Conference on Technological Advances of Thin Films & Surface Coatings
work page 2013
-
[70]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Sca...
work page 2009
- [71]
-
[72]
The Journal of Chemical Physics , volume =
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A consistent and accurateab ini- tioparametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu, The Journal of Chemical Physics132, 10.1063/1.3382344 (2010)
-
[73]
V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey, and J.-L. Br´ edas, Charge transport in organic semiconductors, Chemical Reviews107, 926 (2007), pMID: 17378615, https://doi.org/10.1021/cr050140x
-
[74]
P. Frondelius, A. Hellman, K. Honkala, H. H¨ akkinen, and H. Gr¨ onbeck, Charging of atoms, clusters, and molecules on metal-supported oxides: A general and long-ranged phenomenon, Phys. Rev. B78, 085426 (2008)
work page 2008
-
[75]
L. Giordano and G. Pacchioni, Oxide films at the nanoscale: New structures, new func- tions, and new materials, Accounts of Chemical Research44, 1244 (2011), pMID: 21805966, https://doi.org/10.1021/ar200139y
-
[76]
M. Oehzelt, N. Koch, and G. Heimel, Organic semiconductor density of states controls the en- ergy level alignment at electrode interfaces, Nature Communications5, 10.1038/ncomms5174 (2014)
- [77]
-
[78]
O. T. Hofmann, P. Rinke, M. Scheffler, and G. Heimel, Integer versus fractional charge transfer at metal(/insulator)/organic interfaces: Cu(/nacl)/tcne, ACS Nano9, 5391–5404 (2015)
work page 2015
-
[79]
K. A. Cochrane, A. Schiffrin, T. S. Roussy, M. Capsoni, and S. A. Burke, Pronounced polarization-induced energy level shifts at boundaries of organic semiconductor nanostruc- tures, Nature Communications6, 10.1038/ncomms9312 (2015)
- [80]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.