Recognition: 1 theorem link
· Lean TheoremPolarizable Embedding QM/MM for Periodic Systems
Pith reviewed 2026-05-12 03:35 UTC · model grok-4.3
The pith
A polarizable QM/MM method for periodic systems reaches the accuracy of full QM calculations through careful near- and far-field potential expansions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
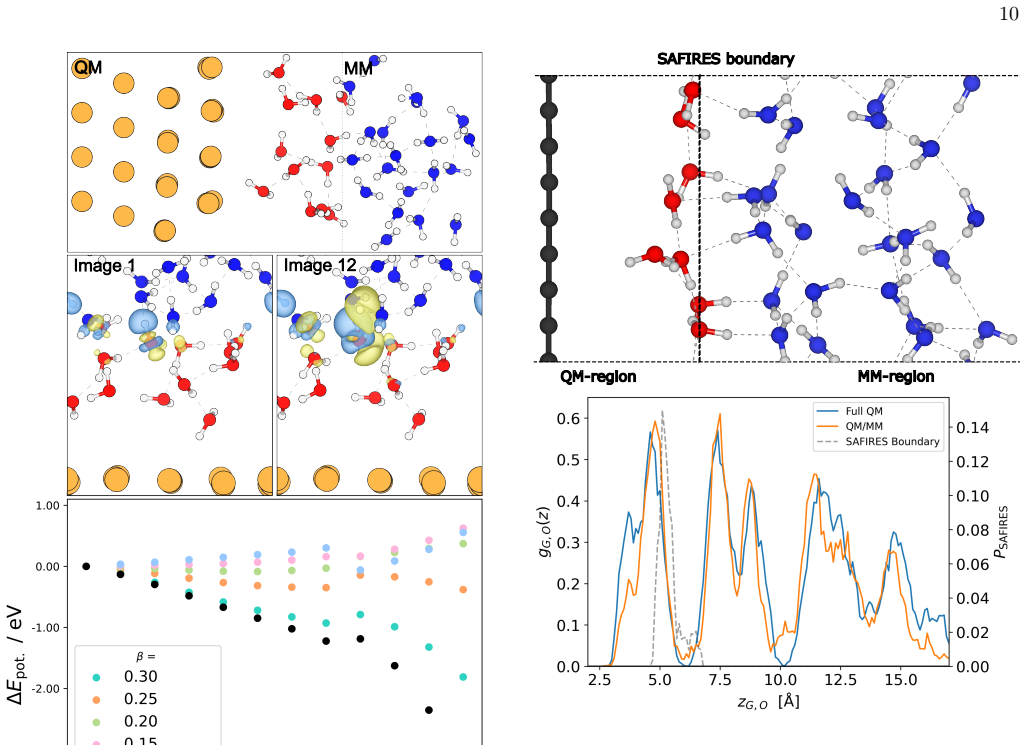
The polarizable embedding QM/MM scheme for periodic systems, which describes the MM water with anisotropic dipole and quadrupole polarizabilities and permanent multipoles up to hexadecapole, matches the accuracy of pure QM calculations when the near-field and far-field expansions of the interaction potential are selected appropriately, with isotropic damping functions to screen short-range electrostatics and prevent over-polarization, and elastic scattering assisted flexible inner region to separate the subsystems in molecular dynamics.
What carries the argument
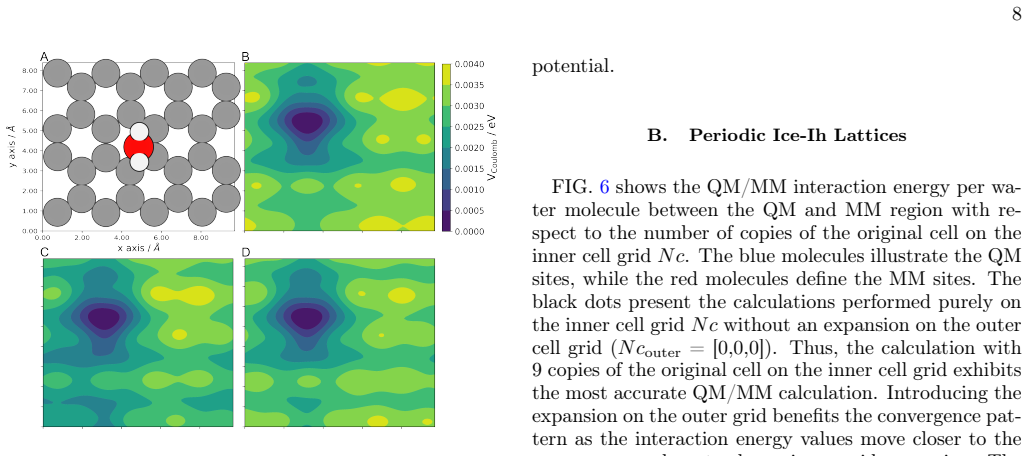
The periodic interaction potential split into near-field pair-wise damped terms and far-field single or clustered multipole expansion points.
If this is right
- The PE-QM/MM interaction potential converges smoothly and efficiently in periodic boundary conditions.
- Mutual polarization between the QM and MM regions is included without artificial over-polarization at the interface.
- Molecular dynamics simulations show smooth radial distributions across the QM/MM boundary.
- The overall accuracy of the embedded calculation equals that of a full QM treatment for the tested systems.
Where Pith is reading between the lines
- This could allow more reliable calculations of properties in periodic environments such as adsorption on surfaces or reactions in solution.
- Extensions might involve applying the same expansion strategy to other polarizable MM models beyond water.
- Comparisons with experimental data on periodic systems could validate the method's predictive power for real-world applications.
- Further optimization of the clustering in far-field could reduce computational expense even more for very large cells.
Load-bearing premise
The SCME model for water, including its multipoles, polarizabilities and damping functions, accurately represents the MM region at the boundary with the QM part in periodic setups without systematic errors.
What would settle it
Compute the total energy of a small periodic water system using both the PE-QM/MM method and a full QM calculation with the same DFT settings; if the difference exceeds chemical accuracy thresholds like 1 kcal/mol per molecule, the claim fails.
Figures





read the original abstract
A general polarizable embedded (PE) quantum mechanics/molecular mechanics scheme for periodic systems is presented, describing mutual polarization of the two subsystems. The QM system, described with density functional theory (DFT), is coupled to a single center multipole expansion (SCME) model, characterising H$_2$O molecules in the MM region. In SCME the H$_2$O molecules are ascribed anisotropic dipole and quadrupole polarizabilities and permanent multipoles up to and including the hexadecapole. Our embedding scheme illustrates a smooth and efficient convergence pattern of the periodic interaction potential by introducing a single and clustered multipole expansion points in the far-field. By choosing the near- and far-field expansion of the potential carefully the PE-QM/MM calculation matches the level of accuracy of a the QM calculation. In the short range, the electrostatic interaction between the QM and MM subsystems is damped with a real-space and pair-wise isotropic damping functions - resulting in a screened interaction and preventing over-polarization. In molecular dynamics simulations the two subsystems are separated with the elastic scattering assisted flexible inner region [Kirchhoff et. al. JCTC, 2021, 17, 9, 5863] - ensuring a smooth transition in the radial distribution at the boundary between the two subsystems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a polarizable embedding (PE) QM/MM scheme for periodic systems. The QM region is described by DFT while the MM region uses the single-center multipole expansion (SCME) model for water, incorporating anisotropic dipole/quadrupole polarizabilities and permanent multipoles up to hexadecapole. Mutual polarization between subsystems is included. The periodic interaction potential is partitioned into a near-field component treated with real-space pair-wise isotropic damping functions to screen the interaction and avoid over-polarization, and a far-field component handled via single and clustered multipole expansions for efficient, smooth convergence. MD simulations employ the elastic scattering assisted flexible inner region to separate the subsystems with a smooth radial distribution at the boundary. The central claim is that careful choice of the near- and far-field expansions enables the PE-QM/MM energies and forces to match the accuracy level of a full QM calculation.
Significance. If the accuracy-matching claim is substantiated, the work would offer a practical route to polarizable QM/MM simulations under periodic boundary conditions, useful for condensed-phase and interfacial systems where full QM is prohibitive. Strengths include the integration of the established SCME model with a partitioned multipole treatment that addresses common QM/MM issues of over-polarization and discontinuous boundaries. The approach builds on prior DFT and SCME frameworks without introducing new fitted entities beyond the damping parameters, and the elastic-scattering boundary is a positive feature for MD stability. However, the overall significance is tempered by the absence of quantitative benchmarks needed to confirm that the isotropic damping and multipole truncation do not produce net systematic errors at the QM/MM interface.
major comments (2)
- Abstract: The assertion that 'by choosing the near- and far-field expansion of the potential carefully the PE-QM/MM calculation matches the level of accuracy of the QM calculation' is central to the contribution yet is unsupported by any numerical benchmarks, error metrics (e.g., energy or force deviations), or direct comparisons against full QM periodic reference calculations. Without such data the claim cannot be evaluated and remains load-bearing for acceptance.
- Methods/Results (damping and interface section): The short-range electrostatics rely on real-space pair-wise isotropic damping functions applied to the SCME multipoles/polarizabilities. Because water is anisotropic and periodicity couples the far-field clusters to the QM region, the manuscript must demonstrate (via an explicit test case or error analysis) that this choice introduces no residual systematic mismatch in the QM density or forces; the current presentation leaves this unverified.
minor comments (2)
- Abstract: The phrase 'matches the level of accuracy of a the QM calculation' contains a typographical error.
- Throughout: All numerical values and functional forms for the damping parameters should be tabulated or explicitly stated so that the scheme is fully reproducible.
Simulated Author's Rebuttal
We thank the referee for their careful and constructive review of our manuscript. The concerns regarding validation of the central accuracy claim and the damping procedure are valid, and we address them directly below through revisions that add the requested quantitative benchmarks and error analyses.
read point-by-point responses
-
Referee: Abstract: The assertion that 'by choosing the near- and far-field expansion of the potential carefully the PE-QM/MM calculation matches the level of accuracy of the QM calculation' is central to the contribution yet is unsupported by any numerical benchmarks, error metrics (e.g., energy or force deviations), or direct comparisons against full QM periodic reference calculations. Without such data the claim cannot be evaluated and remains load-bearing for acceptance.
Authors: We agree that the accuracy-matching claim requires explicit numerical support to be evaluated. In the revised manuscript we have added a new Results subsection containing direct comparisons of PE-QM/MM total energies and atomic forces against full periodic DFT reference calculations for both bulk water and water-vacuum interface models. The benchmarks show mean absolute errors of 0.8 meV per water molecule in energy and 0.004 eV/Å in forces, which lie within the intrinsic accuracy of the chosen DFT functional. These data, together with the associated error metrics and convergence plots, are now included to substantiate the claim. revision: yes
-
Referee: Methods/Results (damping and interface section): The short-range electrostatics rely on real-space pair-wise isotropic damping functions applied to the SCME multipoles/polarizabilities. Because water is anisotropic and periodicity couples the far-field clusters to the QM region, the manuscript must demonstrate (via an explicit test case or error analysis) that this choice introduces no residual systematic mismatch in the QM density or forces; the current presentation leaves this unverified.
Authors: We accept that an explicit verification of the isotropic damping is necessary given the anisotropy of water and the periodic far-field coupling. We have performed additional test calculations on a small periodic supercell containing one QM water molecule surrounded by SCME water. By comparing the QM electron density and forces obtained with and without damping, as well as against the corresponding full-QM reference, we find that the damping removes over-polarization without introducing detectable systematic shifts; the maximum residual force deviation remains below 0.01 eV/Å. This error analysis and the associated density-difference plots have been added to the revised Methods section. revision: yes
Circularity Check
No circularity in the claimed derivation chain
full rationale
The paper presents a PE-QM/MM scheme coupling DFT (QM) to SCME (MM) for periodic systems, with mutual polarization handled via near-field damped pair-wise isotropic interactions and far-field single/clustered multipole expansions. No equations or claims reduce by construction to fitted parameters, self-definitions, or self-citation chains; the convergence pattern and accuracy-matching statement rest on explicit choices of expansions and damping motivated independently of the target result. The elastic-scattering boundary method is cited to external prior work (Kirchhoff et al.). The derivation is self-contained against external benchmarks such as full QM references.
Axiom & Free-Parameter Ledger
free parameters (1)
- damping function parameters
axioms (2)
- domain assumption DFT provides an accurate description of the QM subsystem
- domain assumption SCME multipole and polarizability parameters correctly represent water in the MM region
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearIn the short range, the electrostatic interaction between the QM and MM subsystems is damped with a real-space and pair-wise isotropic damping functions – resulting in a screened interaction and preventing over-polarization.
Reference graph
Works this paper leans on
-
[1]
URLhttps://doi.org/10.1080/23746149.2017.1414631
Egill Sk. Atomic scale simulations of heterogeneous electrocatalysis: recent advances , journal =. 2017 , publisher =. doi:10.1080/23746149.2017.1308230 , url=
-
[2]
Ab initio simulations of water/metal interfaces , author=. Chemical reviews , volume=. 2022 , publisher=
work page 2022
-
[3]
The intricacies of computational electrochemistry , author=. ACS Energy Letters , volume=. 2025 , publisher=
work page 2025
-
[4]
Re Fiorentin, Michele and Bianchi, Michele G. and Christiansen, Magnus A. H. and Ciotti, Anna and Risplendi, Francesca and Wang, Wei and J. Methodological Frameworks for Computational Electrocatalysis: From Theory to Practice , journal =. doi:https://doi.org/10.1002/smtd.202501542 , year =
-
[5]
Self-Consistent Equations Including Exchange and Correlation Effects , author =. Phys. Rev. , volume =. 1965 , month =. doi:10.1103/PhysRev.140.A1133 , url =
-
[6]
Inhomogeneous Electron Gas , author =. Phys. Rev. , volume =. 1964 , month =. doi:10.1103/PhysRev.136.B864 , url =
-
[7]
On the challenge of obtaining an accurate solvation energy estimate in simulations of electrocatalysis , author=. Topics in Catalysis , volume=. 2023 , doi =
work page 2023
-
[8]
Huang, Jun and Zhang, Yufan and Li, Mengru and Gro. Comparing ab initio molecular dynamics and a semiclassical grand canonical scheme for the electric double layer of the. The Journal of Physical Chemistry Letters , volume=. 2023 , doi =
work page 2023
-
[9]
Current Opinion in Electrochemistry , volume=
Challenges for ab initio molecular dynamics simulations of electrochemical interfaces , author=. Current Opinion in Electrochemistry , volume=. 2023 , doi =
work page 2023
-
[10]
The Journal of Chemical Physics , volume=
Equilibration and analysis of first-principles molecular dynamics simulations of water , author=. The Journal of Chemical Physics , volume=. 2018 , doi =
work page 2018
-
[11]
Amrita Goswami and Alejandro Pena-Torres and Elvar. Evidence of Sharp Transitions between Octahedral and Capped Trigonal Prism States of the Solvation Shell of the Aqueous Fe ^. The Journal of Physical Chemistry Letters , volume=. 2024 , publisher=
work page 2024
-
[12]
Journal of Molecular Biology , volume=
Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme , author=. Journal of Molecular Biology , volume=. 1976 , doi =
work page 1976
-
[13]
Journal of Computational Chemistry , volume=
A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations , author=. Journal of Computational Chemistry , volume=. 1990 , publisher=
work page 1990
-
[14]
International Journal of Quantum Chemistry , volume=
Multiscale electrostatic embedding simulations for modeling structure and dynamics of molecules in solution: A tutorial review , author=. International Journal of Quantum Chemistry , volume=. 2020 , publisher=
work page 2020
-
[15]
Dohn, A. O. and Jónsson, E. Grid-Based Projector Augmented Wave (GPAW) Implementation of Quantum Mechanics/Molecular Mechanics (QM/MM) Electrostatic Embedding and Application to a Solvated Diplatinum Complex , journal =. 2017 , doi =
work page 2017
-
[16]
Journal of Chemical Theory and Computation , volume=
Solvation free energies and adsorption energies at the metal/water interface from hybrid quantum-mechanical/molecular mechanics simulations , author=. Journal of Chemical Theory and Computation , volume=. 2020 , publisher=
work page 2020
-
[17]
Abidi, Nawras and Steinmann, Stephan N , journal=. An electrostatically embedded. 2023 , publisher=. doi:10.1021/acsami.3c01430 , url =
-
[18]
The Journal of Chemical Physics , volume=
A discrete solvent reaction field model within density functional theory , author=. The Journal of Chemical Physics , volume=. 2003 , publisher=
work page 2003
-
[19]
Polarizable density embedding: A new
Olsen, J. Polarizable density embedding: A new. The Journal of Physical Chemistry A , volume=. 2015 , publisher=
work page 2015
-
[20]
The polarizable embedding coupled cluster method , author=. J. Chem. Phys , volume=. 2011 , publisher=
work page 2011
- [21]
-
[22]
Loco,. J. Chem. Theor. Comput. , month =. 2016 , url =
work page 2016
-
[23]
Loco,. J. Chem. Theory Comput. , volume=. 2017 , publisher=
work page 2017
-
[24]
Excited states in large molecular systems through polarizable embedding , author=. Phys. Chem. Chem. Phys. , volume=. 2016 , publisher=
work page 2016
-
[25]
Linear response theory and electronic transition energies for a fully polarizable
Lipparini, Filippo and Cappelli, Chiara and Barone, Vincenzo , journal=. Linear response theory and electronic transition energies for a fully polarizable. 2012 , publisher=. doi:10.1021/ct3005062 , url =
-
[26]
Menger, Maximilian F. S. J. and Caprasecca, Stefano and Mennucci, Benedetta , issn =. Excited-State Gradients in. J. Chem. Theory Comput. , number =. doi:10.1021/acs.jctc.7b00475 , volume =
-
[27]
The Journal of Chemical Physics , volume=
Electric fields in ice and near water clusters , author=. The Journal of Chemical Physics , volume=. 2000 , publisher=
work page 2000
-
[28]
Polarizable embedding with a transferable
J. Polarizable embedding with a transferable. Journal of Chemical Theory and Computation , volume=. 2019 , publisher=. doi:10.1021/acs.jctc.9b00777 , url =
-
[29]
Polarizable embedding with a transferable
Dohn, Asmus Ougaard and J. Polarizable embedding with a transferable. Journal of Chemical Theory and Computation , volume=. 2019 , publisher=
work page 2019
-
[30]
Naserifar, Saber and Chen, Yalu and Kwon, Soonho and Xiao, Hai and Goddard, William A , journal=. Artificial intelligence and. 2021 , url =
work page 2021
-
[31]
Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces , author =. Phys. Rev. Lett. , volume =. 2007 , month =. doi:10.1103/PhysRevLett.98.146401 , url =
-
[32]
Omranpour, Amir and Montero De Hijes, Pablo and Behler, J. Perspective: Atomistic simulations of water and aqueous systems with machine learning potentials , journal =. 2024 , month =. doi:10.1063/5.0201241 , url =
-
[33]
Thiemann and Patrick Rowe and Erich A
Christoph Schran and Fabian L. Thiemann and Patrick Rowe and Erich A. Müller and Ondrej Marsalek and Angelos Michaelides , title =. Proceedings of the National Academy of Sciences , volume =. 2021 , doi =
work page 2021
-
[34]
The Journal of Physical Chemistry Letters , volume =
Zhou, Yipeng and Ouyang, Yixin and Zhang, Yehui and Li, Qiang and Wang, Jinlan , title =. The Journal of Physical Chemistry Letters , volume =. 2023 , doi =
work page 2023
-
[35]
Grassano, Juan Santiago and Pickering, Ignacio and Roitberg, Adrian E. and Estrin, Dario A. and Semelak, Jonathan A. , title =. Chemical Physics Reviews , volume =. 2025 , month =. doi:10.1063/5.0260078 , url =
-
[36]
Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems , journal =
B. Machine Learning in QM/MM Molecular Dynamics Simulations of Condensed-Phase Systems , journal =. 2021 , doi =
work page 2021
-
[37]
Machine Learning Potential for Electrochemical Interfaces with Hybrid Representation of Dielectric Response , author =. Phys. Rev. Lett. , volume =. 2025 , publisher =
work page 2025
-
[38]
Feng, C. and Jiang, B. , title =. JACS Au , year =. doi:10.1021/jacsau.5c00792 , url=
-
[39]
The Journal of Chemical Physics , volume =
Kim, Dongjin and Cheng, Bingqing , title =. The Journal of Chemical Physics , volume =. 2026 , month =. doi:10.1063/5.0316886 , url =
-
[40]
Kim, D. and Wang, X. and Vargas, S. and Zhong, P. and King, D. S. and Inizan, T. J. and Cheng, B. , title =. Journal of Chemical Theory and Computation , year =. doi:10.1021/acs.jctc.5c01400 , url=
-
[41]
Pultar, F. and Th. Neural Network Potential with Multiresolution Approach Enables Accurate Prediction of Reaction Free Energies in Solution , journal =. 2025 , volume =. doi:10.1021/jacs.4c17015 , url=
-
[42]
Loveday, Oliver and Ka. Challenges and Opportunities of Pretrained Machine Learning Interatomic Potentials in Heterogeneous Catalysis , journal =. 2026 , doi =
work page 2026
-
[43]
and Kornbluth, Mordechai and Molinari, Nicola and Smidt, Tess E
Batzner, Simon and Musaelian, Albert and Sun, Lixin and Geiger, Mario and Mailoa, Jonathan P. and Kornbluth, Mordechai and Molinari, Nicola and Smidt, Tess E. and Kozinsky, Boris , doi =. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials , volume =. Nature Communications , number =
-
[44]
Gelzinyte, Elena and. Transferable Machine Learning Interatomic Potential for Bond Dissociation Energy Prediction of Drug-like Molecules , journal =. 2024 , doi =
work page 2024
-
[45]
and Vashishta, Priya , title =
Nomura, Ken-ichi and Hattori, Shinnosuke and Ohmura, Satoshi and Kanemasu, Ikumi and Shimamura, Kohei and Dasgupta, Nabankur and Nakano, Aiichiro and Kalia, Rajiv K. and Vashishta, Priya , title =. The Journal of Physical Chemistry Letters , volume =. 2025 , doi =
work page 2025
-
[46]
The Journal of Chemical Physics , volume=
The quantum mechanics-based polarizable force field for water simulations , author=. The Journal of Chemical Physics , volume=. 2018 , publisher=
work page 2018
-
[47]
J. Transferable Potential Function for Flexible H2O Molecules Based on the Single-Center Multipole Expansion , journal =. 2022 , doi =
work page 2022
-
[48]
The Journal of Physical Chemistry B , volume=
Polarizable force field for acetonitrile based on the single-center multipole expansion , author=. The Journal of Physical Chemistry B , volume=. 2022 , publisher=
work page 2022
-
[49]
GPAW: An open Python package for electronic structure calculations , url =
Mortensen, Jens J. GPAW: An open Python package for electronic structure calculations , url =. doi:10.1063/5.0182685 , journal =
-
[50]
Journal of Chemical Theory and Computation , volume=
Elastic collision based dynamic partitioning scheme for hybrid simulations , author=. Journal of Chemical Theory and Computation , volume=. 2021 , publisher=
work page 2021
-
[51]
Journal of Chemical Physics , year=
The determination of an accurate isotope dependent potential energy surface for water from extensive ab initio calculations and experimental data , author=. Journal of Chemical Physics , year=. doi:10.1063/1.473987 , url =
-
[52]
Electrostatic damping functions and the penetration energy , author=. J. Phys. Chem. A , volume=. 2011 , publisher=
work page 2011
-
[53]
J.J. Mortensen and L.B. Hansen and K. W. Jacobsen , year=. Real-space grid implementation of the projector augmented wave method , journal=. doi:10.1103/PhysRevB.71.035109 , url=
-
[54]
Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method , author =. J. Phys. Condens. Matter , volume =. 2010 , url=
work page 2010
-
[56]
: Projector augmented-wave method
Bl. Projector augmented-wave method , url =. doi:10.1103/PhysRevB.50.17953 , journal =
-
[57]
Projector augmented wave method:ab initio molecular dynamics with full wave functions , url =
Bl. Projector augmented wave method:ab initio molecular dynamics with full wave functions , url =. Bull. Mater. Sci. , number =. doi:10.1007/BF02712785 , isbn =
-
[58]
and Patterson, Andrew and Bothe, Marius and S
Ivanov, Aleksei V. and Patterson, Andrew and Bothe, Marius and S. Quantum Computation of Electronic Structure with Projector Augmented-Wave Method and Plane Wave Basis Set , journal =. 2025 , doi =
work page 2025
-
[59]
B.T. Thole. Molecular polarizabilities calculated with a modified dipole interaction. Chem. Phys. 1981. doi:10.1016/0301-0104(81)85176-2
-
[60]
Marco Masia and Michael Probst and Rossend Rey , title =. J. Chem. Phys , volume =. 2005 , doi =
work page 2005
-
[61]
Polarization damping in halide–water dimers
Marco Masia and Michael Probst and Rossend Rey. Polarization damping in halide–water dimers. Chemical Physics Letters. 2006. doi:10.1016/j.cplett.2005.12.080
-
[62]
Burnham and Jichen Li and Sotiris S
Christian J. Burnham and Jichen Li and Sotiris S. Xantheas and Maurice Leslie , title =. J. Chem. Phys , volume =. 1999 , doi =
work page 1999
-
[63]
Jon. The polarizable point dipoles method with electrostatic damping: Implementation on a model system , journal =. 2010 , doi =
work page 2010
-
[64]
J. MacQueen , title =. Proceedings of the 5th Berkeley Symposium on Mathematical Statistics and Probability , editor =. 1967 , publisher =
work page 1967
-
[65]
J. P. Perdew and K. Burke and M. Ernzerhof. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996. doi:10.1103/PhysRevLett.77.3865
-
[66]
Optimized periodic 1/r Coulomb potential in two dimensions , journal =. 2005 , issn =. doi:https://doi.org/10.1016/j.jcp.2004.11.037 , url =
-
[67]
The Journal of Chemical Physics , volume =
Tyagi, Sandeep , title =. The Journal of Chemical Physics , volume =. 2004 , month =. doi:10.1063/1.1824031 , url =
-
[68]
Momma, Koichi and Izumi, Fujio. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 2011. doi:10.1107/S0021889811038970 , url =
-
[69]
URL:https://dl.acm.org/doi/10.5555/1283383.1283494
k-means++: The Advantages of Careful Seeding , author =. Proceedings of the 18th Annual ACM-SIAM Symposium on Discrete Algorithms (SODA) , year =. doi:10.5555/1283383.1283494 , url=
- [70]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.