Recognition: no theorem link
QT-Net: Rethinking Evaluation of AI Models in Atomic Chemical Space
Pith reviewed 2026-05-12 04:02 UTC · model grok-4.3
The pith
QT-Net predicts atomic properties for unseen environments and uses them to improve molecular forecasts.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
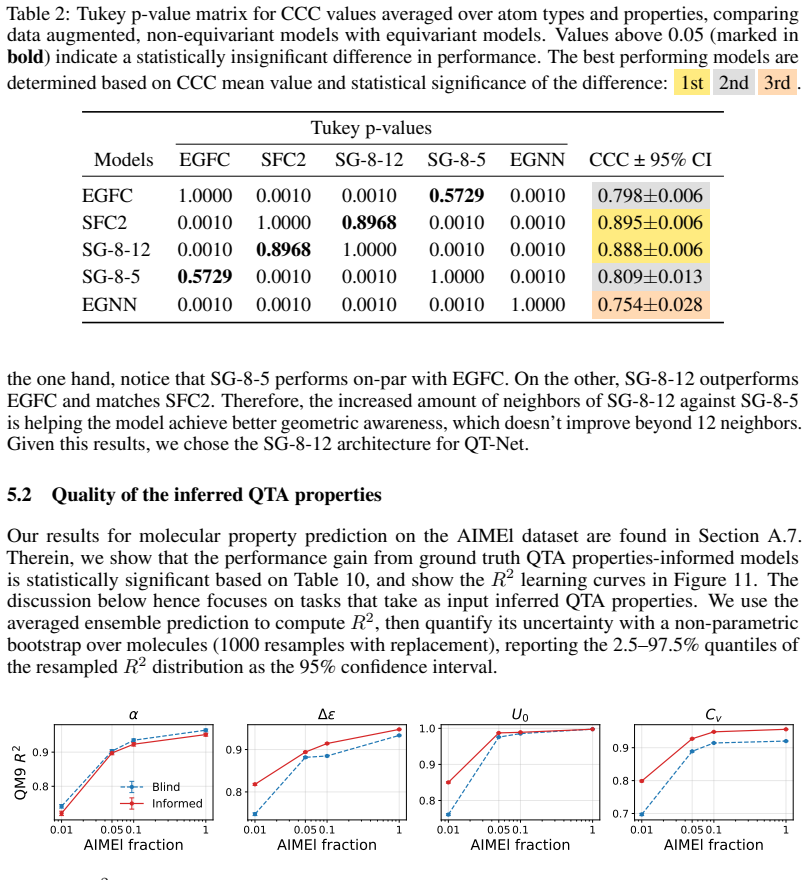
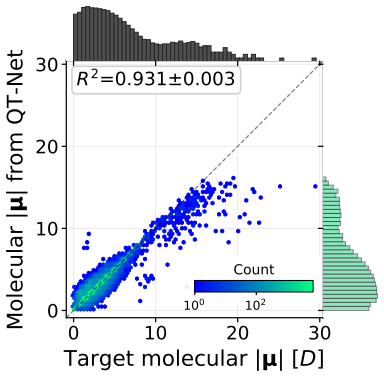
QT-Net, a rotationally augmented non-equivariant graph neural network, is shown to predict electron populations and multipoles for atoms in molecules from QM9 that lie outside the training clusters. These per-atom inferences, when fed as features into other models, improve performance on molecular property prediction tasks. Additionally, summing the atomic contributions from QT-Net recovers the ground-truth molecular dipole moments reported in the QM9 dataset.
What carries the argument
QT-Net, a rotationally augmented non-equivariant graph neural network that outputs per-atom electron populations and multipoles.
If this is right
- QT-Net inferred atomic properties serve as useful input features that enhance downstream molecular property prediction.
- Molecular dipole moments computed by aggregating QT-Net's atomic outputs match the ground-truth values in QM9.
- The SOAP clustering protocol enables rigorous out-of-distribution testing at the atomic level for model comparisons.
- Rotationally augmented non-equivariant models perform competitively with equivariant ones under this evaluation.
Where Pith is reading between the lines
- This suggests that atomic-scale predictions can act as chemical priors to reduce data requirements in broader molecular modeling.
- Applying the same clustering evaluation to other datasets could reveal limitations in current atomic property predictors.
- QT-Net's approach might extend to predicting other atomic-level quantities like energies or forces for simulation tasks.
Load-bearing premise
That grouping atomic environments by SOAP descriptors creates a meaningful out-of-distribution test where unseen clusters represent genuinely novel atomic settings.
What would settle it
A calculation where molecular dipole moments derived from QT-Net per-atom outputs deviate substantially from QM9 ground truth, or where adding QT-Net features fails to improve molecular property prediction accuracy.
Figures














read the original abstract
Atomic properties such as partial charges or multipoles encode chemically meaningful information that can inform downstream molecular property prediction, but their evaluation as machine learning targets has been complicated by the absence of a principled out-of-distribution evaluation protocol at the atomic level. In this work, we propose a held-out evaluation protocol that clusters atomic environments by SOAP descriptors and computes metrics accounting only for cluster labels unseen during training. Following this procedure, we use 5$\times$5 cross-validation and Tukey's HSD to run a statistically rigorous comparison of E(3)-equivariant against non-equivariant, rotationally augmented models for predicting electron populations and multipoles of H, C, N, and O atoms. Building on our results, we introduce the Quantum Topological Neural Network (QT-Net), a rotationally augmented, non-equivariant graph neural network. We show that QT-Net can be used to infer properties of atoms in molecules from QM9 outside our training set, and that these inferred properties can yield improvement when used as input features for downstream molecular property prediction. To further validate the framework, molecular dipole moments computed from QT-Net's per-atom outputs recover the ground-truth values reported in QM9. We release all code and data, including a JAX implementation of QT-Net, to support the broader use of learned QTA properties as inductive biases for atomic-scale molecular machine learning.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes a held-out evaluation protocol for atomic property prediction models that clusters per-atom SOAP descriptors and evaluates only on unseen cluster labels. Using 5×5 cross-validation and Tukey's HSD tests, it compares E(3)-equivariant and non-equivariant (rotationally augmented) models for predicting electron populations and multipoles of H, C, N, O atoms in QM9. It introduces QT-Net, a rotationally augmented non-equivariant GNN, shows that its inferences on atoms from molecules outside the training clusters improve downstream molecular property prediction, and validates the framework by recovering ground-truth QM9 dipole moments from the per-atom outputs. All code and data, including a JAX implementation, are released.
Significance. If the central claims hold, the work is significant for introducing a statistically grounded atomic-level OOD protocol that moves beyond molecule-level splits, for demonstrating that learned QTA properties can serve as useful inductive biases, and for the independent dipole-moment recovery check. The open release of code and data is a clear strength that supports reproducibility and broader adoption in atomic-scale molecular ML.
major comments (2)
- [Evaluation protocol and clustering procedure] The held-out protocol (described in the evaluation section) clusters atomic environments by SOAP descriptors and treats unseen cluster labels as OOD. However, because SOAP is a strictly local descriptor, atoms in chemically similar bonding motifs (e.g., sp2 carbons across different QM9 molecules) can receive high similarity scores yet be placed in different clusters. This risks chemical similarity leakage across the train/test boundary and undermines the claim that QT-Net inferences are performed on genuinely unseen atomic environments. The paper should add quantitative checks (e.g., inter-cluster chemical diversity metrics or molecule-level blocking) to confirm that the splits achieve the intended separation.
- [Downstream molecular property prediction experiments] The downstream improvement claim (reported in the results on molecular property prediction) relies on the OOD inferences being meaningful. If the clustering leakage concern is not addressed, the reported gains when using QT-Net outputs as input features cannot be confidently attributed to true generalization rather than residual training-set correlation. An ablation that compares against a random-cluster baseline or a molecule-level split would strengthen this result.
minor comments (2)
- [Dipole moment validation] The abstract states that dipole moments are recovered from per-atom outputs, but the corresponding results section would benefit from an explicit equation showing how the vector sum is formed and any assumptions about charge neutrality.
- [Background on atomic properties] Notation for the electron population and multipole targets is introduced without a clear reference to the underlying QTAIM definitions; adding a short paragraph or citation in the background section would improve accessibility.
Simulated Author's Rebuttal
Thank you for the opportunity to respond to the referee's report. We appreciate the constructive feedback on our evaluation protocol and downstream experiments. Below, we provide point-by-point responses to the major comments and indicate the revisions we plan to incorporate.
read point-by-point responses
-
Referee: [Evaluation protocol and clustering procedure] The held-out protocol (described in the evaluation section) clusters atomic environments by SOAP descriptors and treats unseen cluster labels as OOD. However, because SOAP is a strictly local descriptor, atoms in chemically similar bonding motifs (e.g., sp2 carbons across different QM9 molecules) can receive high similarity scores yet be placed in different clusters. This risks chemical similarity leakage across the train/test boundary and undermines the claim that QT-Net inferences are performed on genuinely unseen atomic environments. The paper should add quantitative checks (e.g., inter-cluster chemical diversity metrics or molecule-level blocking) to confirm that the splits achieve the intended separation.
Authors: We agree that SOAP descriptors are local and that clustering algorithms may place chemically related atoms into separate clusters when their descriptor vectors differ sufficiently. The protocol defines OOD strictly via unseen cluster labels rather than claiming perfect chemical isolation. To address the concern directly, we will add quantitative checks in the revised manuscript: average inter-cluster SOAP cosine similarities, distributions of bond orders and atomic coordination numbers across clusters, and an analysis of molecule-level overlap. These metrics will quantify the achieved separation and support the claim that test atoms represent environments outside the training distribution in descriptor space. revision: yes
-
Referee: [Downstream molecular property prediction experiments] The downstream improvement claim (reported in the results on molecular property prediction) relies on the OOD inferences being meaningful. If the clustering leakage concern is not addressed, the reported gains when using QT-Net outputs as input features cannot be confidently attributed to true generalization rather than residual training-set correlation. An ablation that compares against a random-cluster baseline or a molecule-level split would strengthen this result.
Authors: We concur that linking downstream gains to true OOD generalization requires additional controls. In the revision we will include an ablation that replaces the SOAP-based clustering with random cluster assignments while preserving the same held-out fraction, allowing direct comparison of whether the descriptor-driven splits produce distinct benefits. We will also report atomic-property prediction and downstream results under a conventional molecule-level train/test split. These experiments will help isolate the contribution of the learned QTA properties from any residual correlations. revision: yes
Circularity Check
No significant circularity in QT-Net derivation
full rationale
The paper defines an atomic-level OOD protocol via SOAP clustering and 5x5 CV, trains QT-Net on electron populations/multipoles, then validates via downstream molecular property gains and independent recovery of QM9 dipole moments from per-atom outputs. These steps rely on external QM9 benchmarks and empirical comparisons rather than reducing to fitted inputs or self-definitions by construction. No load-bearing self-citations, no ansatz smuggling, and no renaming of known results appear in the provided text. The central claims remain self-contained against external data.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Javier E Alfonso-Ramos, Rebecca M Neeser, and Thijs Stuyver. Repurposing quantum chemical descriptor datasets for on-the-fly generation of informative reaction representations: Application to hydrogen atom transfer reactions.Digital Discovery, 3(5):919–931, 2024
work page 2024
-
[2]
Evan R Antoniuk, Shehtab Zaman, Tal Ben-Nun, Peggy Li, James Diffenderfer, Busra Sahin, Obadiah Smolenski, Tim Hsu, Anna M Hiszpanski, Kenneth Chiu, et al. BOOM: Benchmarking out-of-distribution molecular property predictions of machine learning models.arXiv preprint arXiv:2505.01912, 2025
-
[3]
Albert P Bartók, Risi Kondor, and Gábor Csányi. On representing chemical environments.Physical Review B—Condensed Matter and Materials Physics, 87(18):184115, 2013
work page 2013
- [4]
-
[5]
Tristan Bereau, Denis Andrienko, and O Anatole V on Lilienfeld. Transferable atomic multipole machine learning models for small organic molecules.Journal of Chemical Theory and Computation, 11(7): 3225–3233, 2015
work page 2015
-
[6]
Cristian Bodnar, Fabrizio Frasca, Nina Otter, Yuguang Wang, Pietro Lio, Guido F Montufar, and Michael Bronstein. Weisfeiler and Lehman go cellular: CW networks.Advances in Neural Information Processing Systems, 34:2625–2640, 2021
work page 2021
-
[7]
Matthew L Brown, Bienfait K Isamura, Jonathan M Skelton, and Paul LA Popelier. Incorporating noncovalent interactions in transfer learning gaussian process regression models for molecular simulations. Journal of Chemical Theory and Computation, 20(14):5994–6008, 2024
work page 2024
-
[8]
Jordan, Marisa Gliege, Santiago Miret, Vijay Kris Narasimhan, and Rocío Mercado
Pablo Martínez Crespo, Robert S. Jordan, Marisa Gliege, Santiago Miret, Vijay Kris Narasimhan, and Rocío Mercado. Topomole: Topological message passing meets hyperedge messages. InAI for Accelerated Materials Design - NeurIPS 2025, 2025. URLhttps://openreview.net/forum?id=k39fE6SD4u
work page 2025
-
[9]
Miguel Gallegos, Jose Manuel Guevara-Vela, and Ángel Martín Pendás. NNAIMQ: A neural network model for predicting QTAIM charges.The Journal of Chemical Physics, 156(1), 2022
work page 2022
-
[10]
Miguel Gallegos, Valentin Vassilev-Galindo, Igor Poltavsky, Ángel Martín Pendás, and Alexandre Tkatchenko. Explainable chemical artificial intelligence from accurate machine learning of real-space chemical descriptors.Nature Communications, 15(1):4345, 2024
work page 2024
-
[11]
Wenhao Gao, Tianfan Fu, Jimeng Sun, and Connor Coley. Sample efficiency matters: A benchmark for practical molecular optimization.Advances in Neural Information Processing Systems, 35:21342–21357, 2022
work page 2022
-
[12]
Johannes Gasteiger, Janek Groß, and Stephan Günnemann. Directional message passing for molecular graphs.arXiv preprint arXiv:2003.03123, 2020
-
[13]
Winston Gee, Abigail Doyle, Santiago Vargas, and Anastassia N Alexandrova. Multi-level qtaim-enriched graph neural networks for resolving properties of transition metal complexes.Digital Discovery, 4(11): 3378–3388, 2025
work page 2025
-
[14]
Yanfei Guan, Connor W Coley, Haoyang Wu, Duminda Ranasinghe, Esther Heid, Thomas J Struble, Lagnajit Pattanaik, William H Green, and Klavs F Jensen. Regio-selectivity prediction with a machine- learned reaction representation and on-the-fly quantum mechanical descriptors.Chemical Science, 12(6): 2198–2208, 2021
work page 2021
-
[15]
Democratising real-world drug discovery through agentic ai.Drug Discovery Today, page 104605, 2026
Jiazhen He, Helen Lai, Lakshidaa Saigiridharan, Gian Marco Ghiandoni, Kinga Jenei, Umur Gokalp, Ajsa Nukovic, Ola Engkvist, Jon Paul Janet, and Samuel Genheden. Democratising real-world drug discovery through agentic ai.Drug Discovery Today, page 104605, 2026
work page 2026
-
[16]
M Hoffmann, A Kazimir, T Oestereich, L Kaermer, F Engelberger, J Meiler, and C Lamers. A quantum lens on molecular design: A machine-learned energy function from interacting quantum atoms.bioRxiv, pages 2026–03, 2026
work page 2026
-
[17]
Ross Irwin, Alessandro Tibo, Jon Paul Janet, and Simon Olsson. SemlaFlow–Efficient 3D molecular generation with latent attention and equivariant flow matching.arXiv preprint arXiv:2406.07266, 2024. 11
-
[18]
Kjell Jorner, Tore Brinck, Per-Ola Norrby, and David Buttar. Machine learning meets mechanistic modelling for accurate prediction of experimental activation energies.Chemical Science, 12(3):1163–1175, 2021
work page 2021
-
[19]
Koichiro Kato, Tomohide Masuda, Chiduru Watanabe, Naoki Miyagawa, Hideo Mizouchi, Shumpei Nagase, Kikuko Kamisaka, Kanji Oshima, Satoshi Ono, Hiroshi Ueda, et al. High-precision atomic charge prediction for protein systems using fragment molecular orbital calculation and machine learning.Journal of Chemical Information and Modeling, 60(7):3361–3368, 2020
work page 2020
-
[20]
Dávid Péter Kovács, J Harry Moore, Nicholas J Browning, Ilyes Batatia, Joshua T Horton, Yixuan Pu, Venkat Kapil, William C Witt, Ioan-Bogdan Magdau, Daniel J Cole, et al. Mace-off: Short-range transferable machine learning force fields for organic molecules.Journal of the American Chemical Society, 147(21):17598–17611, 2025
work page 2025
-
[21]
Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G
Daniel S Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G Taylor, Muhammad R Hasyim, Kyle Michel, Ilyes Batatia, Gábor Csányi, Misko Dzamba, Peter Eastman, et al. The Open Molecules 2025 (OMol25) dataset, evaluations, and models.arXiv preprint arXiv:2505.08762, 2025
-
[22]
Sarah Lewis, Tim Hempel, José Jiménez-Luna, Michael Gastegger, Yu Xie, Andrew YK Foong, Victor Gar- cía Satorras, Osama Abdin, Bastiaan S Veeling, Iryna Zaporozhets, et al. Scalable emulation of protein equilibrium ensembles with generative deep learning.Science, 389(6761):eadv9817, 2025
work page 2025
-
[23]
Shih-Cheng Li, Haoyang Wu, Angiras Menon, Kevin A Spiekermann, Yi-Pei Li, and William H Green. When do quantum mechanical descriptors help graph neural networks to predict chemical properties? Journal of the American Chemical Society, 146(33):23103–23120, 2024
work page 2024
-
[24]
Chérif F Matta and Russell J Boyd. An introduction to the quantum theory of atoms in molecules.The quantum theory of atoms in molecules, 1, 2007
work page 2007
-
[25]
Peter Maxwell, Nicodemo di Pasquale, Salvatore Cardamone, and Paul LA Popelier. The prediction of topologically partitioned intra-atomic and inter-atomic energies by the machine learning method kriging. Theoretical Chemistry Accounts, 135(8):195, 2016
work page 2016
-
[26]
hdbscan: Hierarchical density based clustering.Journal of Open Source Software, 2(11):205, 2017
Leland McInnes, John Healy, Steve Astels, et al. hdbscan: Hierarchical density based clustering.Journal of Open Source Software, 2(11):205, 2017
work page 2017
-
[27]
Derek P Metcalf, Andy Jiang, Steven A Spronk, Daniel L Cheney, and C David Sherrill. Electron-passing neural networks for atomic charge prediction in systems with arbitrary molecular charge.Journal of Chemical Information and Modeling, 61(1):115–122, 2020
work page 2020
-
[28]
Quantum topological atomic properties of 44k molecules.Scientific Data, 11(1):945, 2024
Brandon Meza-González, David I Ramírez-Palma, Pablo Carpio-Martínez, David Vázquez-Cuevas, Ka- rina Martínez-Mayorga, and Fernando Cortés-Guzmán. Quantum topological atomic properties of 44k molecules.Scientific Data, 11(1):945, 2024
work page 2024
-
[29]
Brandon Meza-González, David I. Ramírez-Palma, Pablo Carpio-Martínez, David Vázquez-Cuevas, Karina Martinez-Mayorga, and Fernando Cortés-Guzmán. Aimel-db: Atomic properties for 44k small organic molecules, February 2024. URLhttps://doi.org/10.5281/zenodo.11406726
-
[30]
Robert G Parr and Weitao Yang. Density functional approach to the frontier-electron theory of chemical reactivity.Journal of the American Chemical Society, 106(14):4049–4050, 1984
work page 1984
-
[31]
Boltz-2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, et al. Boltz-2: Towards accurate and efficient binding affinity prediction.bioRxiv, 2025
work page 2025
-
[32]
Sergey Pozdnyakov and Michele Ceriotti. Smooth, exact rotational symmetrization for deep learning on point clouds.Advances in Neural Information Processing Systems, 36:79469–79501, 2023
work page 2023
-
[33]
Quantum chemistry structures and properties of 134 kilo molecules.Scientific Data, 1(1):1–7, 2014
Raghunathan Ramakrishnan, Pavlo O Dral, Matthias Rupp, and O Anatole V on Lilienfeld. Quantum chemistry structures and properties of 134 kilo molecules.Scientific Data, 1(1):1–7, 2014
work page 2014
-
[34]
Equivariant message passing for the prediction of tensorial properties and molecular spectra
Kristof Schütt, Oliver Unke, and Michael Gastegger. Equivariant message passing for the prediction of tensorial properties and molecular spectra. InInternational Conference on Machine Learning, pages 9377–9388. PMLR, 2021
work page 2021
-
[35]
Have protein-ligand co-folding methods moved beyond memorisation?bioRxiv, pages 2025–02, 2025
Peter Škrinjar, Jérôme Eberhardt, Janani Durairaj, and Torsten Schwede. Have protein-ligand co-folding methods moved beyond memorisation?bioRxiv, pages 2025–02, 2025. 12
work page 2025
-
[36]
Thijs Stuyver and Connor W Coley. Quantum chemistry-augmented neural networks for reactivity prediction: Performance, generalizability, and explainability.The Journal of Chemical Physics, 156(8), 2022
work page 2022
-
[37]
Santiago Vargas, Winston Gee, and Anastassia Alexandrova. High-throughput quantum theory of atoms in molecules (QTAIM) for geometric deep learning of molecular and reaction properties.Digital Discovery, 3(5):987–998, 2024
work page 2024
-
[38]
Max Veit, David M Wilkins, Yang Yang, Robert A DiStasio, and Michele Ceriotti. Predicting molecular dipole moments by combining atomic partial charges and atomic dipoles.The Journal of chemical physics, 153(2), 2020
work page 2020
-
[39]
Jike Wang, Dongsheng Cao, Cunchen Tang, Lei Xu, Qiaojun He, Bo Yang, Xi Chen, Huiyong Sun, and Tingjun Hou. DeepAtomicCharge: A new graph convolutional network-based architecture for accurate prediction of atomic charges.Briefings in Bioinformatics, 22(3):bbaa183, 2021
work page 2021
-
[40]
A generative model for inorganic materials design.Nature, 639(8055):624–632, 2025
Claudio Zeni, Robert Pinsler, Daniel Zügner, Andrew Fowler, Matthew Horton, Xiang Fu, Zilong Wang, Aliaksandra Shysheya, Jonathan Crabbé, Shoko Ueda, et al. A generative model for inorganic materials design.Nature, 639(8055):624–632, 2025. 13 A Technical Appendices and Supplementary Material A.1 Training and computational details We report training and co...
work page 2025
-
[41]
— arise naturally when training-set variability is dominated by which environments areexcluded rather than included, and do not indicate a violation. The effective sample size neff ∈[16.5,25.0] across models reflects this near-independence and confirms that the standard repeated-CV estimator remains close to unbiased. Normality and homoscedasticity (SW an...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.