Recognition: 2 theorem links
· Lean TheoremMicro-environment of the Eu interstitial in β-SiAlON:Eu²⁺ green phosphor
Pith reviewed 2026-05-12 04:11 UTC · model grok-4.3
The pith
First-principles calculations reproduce the low-temperature photoluminescence spectrum of beta-SiAlON:Eu2+ and validate the Eu-N9 coordination model.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
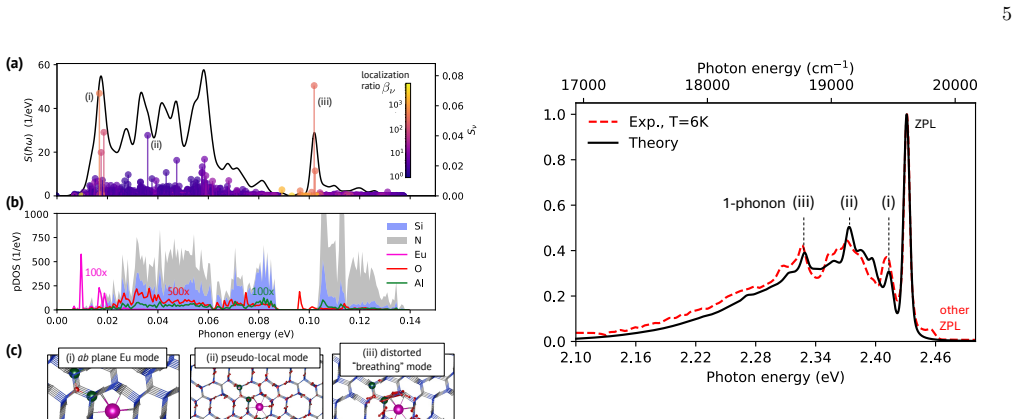
Using the first-principles DeltaSCF excited-state method, embedding of interatomic force constants for supercells up to 3501 atoms, and Huang-Rhys theory, the authors identify representative low-energy structural models for beta-Si6-zAlzOzN8-z:Eu2+. For the lowest-energy structure at low z, the computed photoluminescence spectrum reproduces the experimental vibronic peaks at 6 K with excellent agreement in both positions and intensities. This agreement validates the Eu-N9 coordination model in which Al, O, and Eu are confined to the same crystallographic plane. Across the low-energy structures the electron-phonon coupling is weak with S approximately 2.15 and a consistent phonon signature, a
What carries the argument
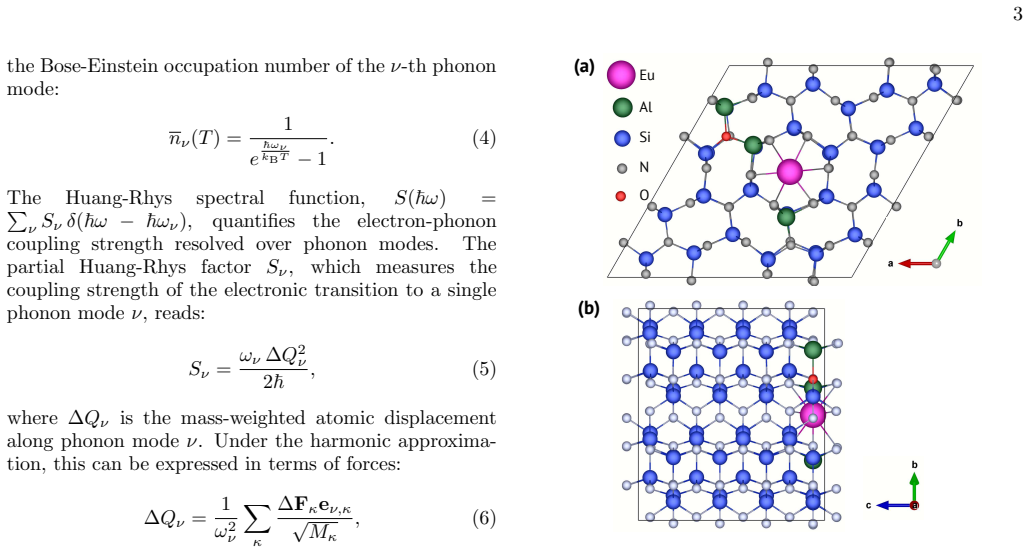
Eu-N9 coordination model with Al, O, and Eu confined to the same crystallographic plane, validated by matching computed and experimental vibronic spectra.
If this is right
- The photoluminescence spectrum including vibronic peak positions and intensities can be predicted accurately from the identified low-energy structures.
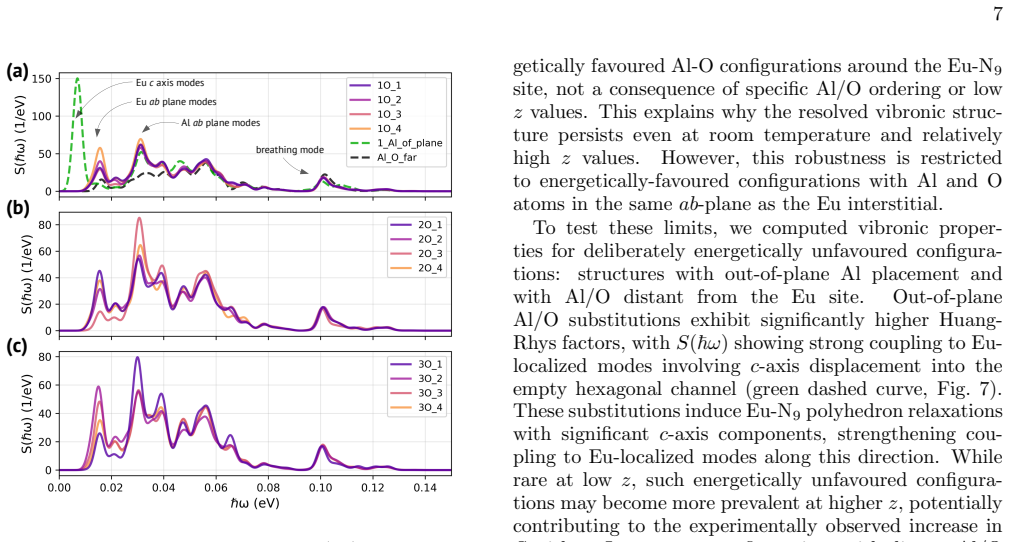
- Electron-phonon coupling strength remains low (S approximately 2.15) and the characteristic phonon signature stays robust regardless of Al/O arrangement.
- Resolved phonon replicas persist with increasing z because of the weak and consistent coupling.
- Emission red-shifts with rising z because zero-phonon line energies decrease systematically while Huang-Rhys factors increase modestly and configurational diversity grows.
Where Pith is reading between the lines
- The plane-confined Eu-N9 arrangement may limit non-radiative pathways and thereby contribute to the high quantum efficiency of the commercial phosphor.
- The robustness of the phonon signature suggests that similar coordination motifs could be engineered in related nitride hosts to maintain narrow emission bands.
- The identified trends in zero-phonon line and coupling strength with z provide a direct route to predict emission color shifts in compositional variants without new experiments.
- Extending the same embedding and Monte Carlo protocol to other rare-earth activators could clarify why some phosphors lose spectral resolution at high dopant concentrations.
Load-bearing premise
The Monte Carlo-sampled lowest-energy structures accurately represent the atomic configurations present in the real material and the DeltaSCF plus embedded-force-constant approach captures the electron-phonon coupling without significant errors from the exchange-correlation functional or finite-size effects.
What would settle it
High-resolution local probe measurements such as EXAFS or aberration-corrected STEM that show a coordination number other than nine or a different relative positioning of Al, O, and Eu atoms would falsify the validated model.
Figures



read the original abstract
The precise atomic-scale structure around Eu$^{2+}$ activators in the $\beta$-Si$_{6-z}$Al$_z$O$_z$N$_{8-z}$:Eu$^{2+}$ commercial green phosphor remains elusive. We use the first-principles $\Delta$SCF excited-state method, embedding of the interatomic force constants for supercells up to 3501 atoms, and Huang-Rhys theory to clarify this issue. Monte Carlo exploration is used to identify representative low-energy structural models spanning different levels of Al/O concentration $z$. For the lowest-energy structure at low $z$, our computed photoluminescence spectrum reproduces the experimental vibronic peaks at 6~K with excellent agreement in peak positions and intensities, validating the Eu-N$_9$ coordination model with Al, O, and Eu confined to the same crystallographic plane. Analysis of the low-energy structures reveals that the electron-phonon coupling is weak ($S \approx 2.15$) with a robust characteristic phonon signature across different Al/O arrangements, explaining the surprising persistence of resolved phonon replicas with increasing $z$. We explain the experimentally observed red-shift of emission with increasing $z$ through systematic trends in zero-phonon line energies, modest increases in Huang-Rhys factors, and larger configurational diversity at higher compositions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper applies first-principles ΔSCF excited-state calculations, embedding of interatomic force constants in supercells up to 3501 atoms, and Huang-Rhys theory, together with Monte Carlo sampling of low-energy Al/O configurations in β-Si_{6-z}Al_zO_zN_{8-z}:Eu^{2+}. For the lowest-energy low-z structure it reports that the computed photoluminescence spectrum reproduces the experimental vibronic peaks at 6 K in both position and intensity, thereby validating an Eu-N_9 coordination model in which Al, O, and Eu lie in the same crystallographic plane. The work further finds weak electron-phonon coupling (S ≈ 2.15) that is robust across sampled structures and attributes the observed red-shift with increasing z to systematic trends in zero-phonon-line energies, modest growth in Huang-Rhys factors, and increased configurational diversity.
Significance. If the structural assignment is confirmed, the study supplies a parameter-free, atomistic account of the local environment and vibronic properties of a commercially relevant green phosphor. The use of large embedded supercells, direct computation of the Huang-Rhys factor rather than fitting, and reproduction of low-temperature spectral features constitute clear methodological strengths that would strengthen the credibility of the proposed micro-environment model and its implications for composition-dependent emission.
major comments (3)
- [Abstract and spectral comparison section] Abstract and the section presenting the spectral comparison: the central validation that the Eu-N_9 coplanar model is correct rests on the match obtained for a single lowest-energy Monte Carlo structure. No control spectra are shown for other low-energy MC samples, alternative Al/O placements, or non-coplanar configurations computed with the identical ΔSCF + embedded-force-constant + Huang-Rhys protocol. Without such tests it remains possible that comparable agreement could be obtained for structurally distinct models, weakening the uniqueness of the structural claim.
- [Methods and results on accuracy] Methods and results sections on accuracy assessment: no uncertainty estimates (error bars, convergence tests) are supplied for the computed zero-phonon-line energies, Huang-Rhys factors, or side-band intensities. Likewise, no comparison is made with alternative exchange-correlation functionals or with smaller/larger supercells to quantify possible systematic shifts in ZPL or S arising from the chosen functional or finite-size effects in the embedding procedure.
- [Analysis of low-energy structures] Section analyzing low-energy structures: the assumption that the Monte Carlo lowest-energy models are representative of the real material at low z is not accompanied by explicit checks on sampling convergence or on the possible spectral contribution of higher-lying configurations that may be thermally accessible.
minor comments (2)
- [Abstract] Abstract: the phrase 'excellent agreement' is used without a quantitative metric (e.g., mean absolute deviation of peak positions or integrated intensity ratios); adding a brief numerical measure would improve clarity.
- [Figure captions and text] Figure captions and text: ensure that all reported S values and ZPL shifts are explicitly linked to the corresponding equations in the Huang-Rhys formalism so that readers can trace the numerical values back to the underlying definitions.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed report. We address each major comment point by point below, providing the strongest honest defense based on the manuscript content while agreeing to revisions that strengthen the work without misrepresentation.
read point-by-point responses
-
Referee: [Abstract and spectral comparison section] Abstract and the section presenting the spectral comparison: the central validation that the Eu-N_9 coplanar model is correct rests on the match obtained for a single lowest-energy Monte Carlo structure. No control spectra are shown for other low-energy MC samples, alternative Al/O placements, or non-coplanar configurations computed with the identical ΔSCF + embedded-force-constant + Huang-Rhys protocol. Without such tests it remains possible that comparable agreement could be obtained for structurally distinct models, weakening the uniqueness of the structural claim.
Authors: We agree that additional control calculations would strengthen the uniqueness of the Eu-N9 coplanar assignment. The manuscript already reports that the Huang-Rhys factor is robust (S ≈ 2.15) across multiple low-energy Monte Carlo structures at low z, with consistent phonon signatures. For the revised version we will add the computed photoluminescence spectra for two additional low-energy MC samples and one alternative Al/O arrangement, all using the identical ΔSCF + embedded-force-constant + Huang-Rhys protocol. These will demonstrate that the quantitative match to the 6 K experimental vibronic peaks (positions and intensities) is best for the lowest-energy coplanar model. Non-coplanar configurations lie at substantially higher energies and are not representative at low z; we will explicitly note this in the revision. revision: yes
-
Referee: [Methods and results on accuracy] Methods and results sections on accuracy assessment: no uncertainty estimates (error bars, convergence tests) are supplied for the computed zero-phonon-line energies, Huang-Rhys factors, or side-band intensities. Likewise, no comparison is made with alternative exchange-correlation functionals or with smaller/larger supercells to quantify possible systematic shifts in ZPL or S arising from the chosen functional or finite-size effects in the embedding procedure.
Authors: We acknowledge that explicit uncertainty quantification and convergence data are not presented in the current manuscript. The calculations used supercells up to 3501 atoms with embedding of force constants to reduce finite-size errors, and a standard ΔSCF implementation. In the revision we will add a dedicated subsection on numerical accuracy that includes (i) convergence of ZPL and S with embedded supercell size for a representative structure, (ii) estimated numerical uncertainties arising from the embedding cutoff and force-constant truncation, and (iii) a brief discussion of the chosen exchange-correlation functional together with the expected direction of systematic shifts based on literature benchmarks for similar Eu2+ systems. Direct comparison with alternative functionals for the full 3500-atom protocol is computationally prohibitive, but we will quantify the effect on a smaller test cell. revision: yes
-
Referee: [Analysis of low-energy structures] Section analyzing low-energy structures: the assumption that the Monte Carlo lowest-energy models are representative of the real material at low z is not accompanied by explicit checks on sampling convergence or on the possible spectral contribution of higher-lying configurations that may be thermally accessible.
Authors: The Monte Carlo procedure was employed to locate the lowest-energy Al/O arrangements at low z; the manuscript focuses on these because they dominate the configurational ensemble. To address the referee’s concern we will expand the methods section with details on the number of independent MC runs, total steps, and acceptance statistics to demonstrate that the reported lowest-energy structure is reproducibly obtained. We will also add a short analysis of the energy distribution of sampled configurations and estimate Boltzmann weights at synthesis and measurement temperatures, showing that higher-lying structures contribute negligibly to the low-temperature spectrum. This will be presented as a new paragraph in the low-energy structures section. revision: yes
Circularity Check
First-principles ΔSCF + Huang-Rhys computation is independent of target spectrum
full rationale
The paper's derivation chain consists of Monte Carlo sampling of low-energy structures in β-SiAlON:Eu²⁺, followed by parameter-free ΔSCF excited-state calculations, embedding of interatomic force constants, and direct application of Huang-Rhys theory to obtain the photoluminescence spectrum and S ≈ 2.15. The computed spectrum is compared to (not fitted to) experimental 6 K vibronic peaks for validation. No equations or steps reduce the output to the input data by construction, no parameters are adjusted to the target spectrum, and no load-bearing self-citation or ansatz is invoked to force the result. The approach remains self-contained against external experimental benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption The ΔSCF method accurately describes the 4f-5d excited state of Eu²⁺ in the SiAlON host without significant self-interaction or relaxation errors.
- domain assumption Embedding force constants from smaller cells into 3501-atom supercells faithfully reproduces the local phonon modes around the Eu site.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We use the first-principles ΔSCF excited-state method, embedding of the interatomic force constants for supercells up to 3501 atoms, and Huang-Rhys theory... The Huang-Rhys spectral function S(ℏω)=∑ν Sν δ(ℏω−ℏων)... Sν=ων ΔQν² / 2ℏ
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
For the lowest-energy structure at low z, our computed photoluminescence spectrum reproduces the experimental vibronic peaks at 6 K with excellent agreement
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
S. Nakamura and G. Fasol,The blue laser diode: GaN based light emitters and lasers(Springer Science & Busi- ness Media, 2013)
work page 2013
-
[2]
R.-J. Xie, N. Hirosaki, and T. Takeda, Wide color gamut backlight for liquid crystal displays using three-band phosphor-converted white light-emitting diodes, Applied physics express2, 022401 (2009)
work page 2009
-
[3]
N. Hirosaki, R.-J. Xie, K. Kimoto, T. Sekiguchi, Y. Ya- mamoto, T. Suehiro, and M. Mitomo, Characterization and properties of green-emittingβ-SiAlON: Eu 2+ pow- der phosphors for white light-emitting diodes, Applied Physics Letters86(2005)
work page 2005
-
[4]
Y. Wang and R. Kang, Narrow-band green phosphors for advanced display technologies: Material design, per- formance optimization, and future prospects, Advanced Optical Materials13, e01162 (2025)
work page 2025
-
[5]
R.-J. Xie, N. Hirosaki, H.-L. Li, Y. Li, and M. Mit- omo, Synthesis and photoluminescence properties ofβ- SiAlON: Eu 2+ (Si6−zAlzOzN8−z: Eu 2+): A promis- ing green oxynitride phosphor for white light-emitting diodes, Journal of the Electrochemical Society154, J314 (2007)
work page 2007
- [6]
-
[7]
K. Kimoto, R.-J. Xie, Y. Matsui, K. Ishizuka, and N. Hi- rosaki, Direct observation of single dopant atom in light- emitting phosphor ofβ-SiAlON: Eu 2+, Applied Physics Letters94(2009)
work page 2009
-
[8]
Z. Wang, W. Ye, I.-H. Chu, and S. P. Ong, Elucidat- ing structure–composition–property relationships of the β-SiAlON: Eu 2+ phosphor, Chemistry of Materials28, 8622 (2016)
work page 2016
-
[9]
C. Cozzan, G. Laurita, M. W. Gaultois, M. Cohen, A. A. Mikhailovsky, M. Balasubramanian, and R. Seshadri, Understanding the links between composition, polyhe- dral distortion, and luminescence properties in green- emittingβ-Si 6−zAlzOzN8−z: Eu 2+ phosphors, Journal of Materials Chemistry C5, 10039 (2017)
work page 2017
- [10]
-
[11]
K. Takahashi, K.-I. Yoshimura, M. Harada, Y. Tomo- mura, T. Takeda, R.-J. Xie, and N. Hirosaki, On the origin of fine structure in the photoluminescence spec- tra of theβ-SiAlON: Eu 2+ green phosphor, Science and Technology of Advanced Materials13, 015004 (2012)
work page 2012
-
[12]
X. Zhang, M.-H. Fang, Y.-T. Tsai, A. Lazarowska, S. Mahlik, T. Lesniewski, M. Grinberg, W. K. Pang, F. Pan, C. Liang,et al., Controlling of structural order- ing and rigidity ofβ-SiAlON: Eu through chemical co- substitution to approach narrow-band-emission for light- emitting diodes application, Chemistry of Materials29, 6781 (2017)
work page 2017
- [13]
-
[14]
O. Gunnarsson and B. I. Lundqvist, Exchange and corre- lation in atoms, molecules, and solids by the spin-density- functional formalism, Physical Review B13, 4274 (1976)
work page 1976
-
[15]
R. O. Jones and O. Gunnarsson, The density functional formalism, its applications and prospects, Reviews of Modern Physics61, 689 (1989)
work page 1989
-
[16]
Y. Jia, A. Miglio, S. Ponc´ e, M. Mikami, and X. Gonze, First-principles study of the luminescence of Eu2+-doped phosphors, Phys. Rev. B96, 125132 (2017)
work page 2017
-
[17]
J. Bouquiaux, S. Ponc´ e, Y. Jia, A. Miglio, M. Mikami, and X. Gonze, Importance of Long-Range Chan- nel Sr Displacements for the Narrow Emission in Sr[Li2Al2O2N2]:Eu2+ Phosphor, Advanced Optical Ma- terials9, 2100649 (2021)
work page 2021
-
[18]
K. Huang and A. Rhys, Theory of light absorption and non-radiative transitions in f-centres, Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences204, 406 (1950)
work page 1950
-
[19]
A. Alkauskas, B. B. Buckley, D. D. Awschalom, and C. G. Van de Walle, First-principles theory of the luminescence lineshape for the triplet transition in diamond NV cen- tres, New J. Phys.16, 073026 (2014)
work page 2014
-
[20]
Y. Jin, M. Govoni, G. Wolfowicz, S. E. Sullivan, F. J. Heremans, D. D. Awschalom, and G. Galli, Photolumi- nescence spectra of point defects in semiconductors: Val- idation of first-principles calculations, Physical Review Materials5, 084603 (2021)
work page 2021
-
[21]
S. Kirkpatrick, C. D. Gelatt Jr, and M. P. Vecchi, Opti- mization by simulated annealing, science220, 671 (1983)
work page 1983
-
[22]
J. J. Cordell, J. Pan, A. C. Tamboli, G. J. Tucker, and S. Lany, Probing configurational disorder in ZnGeN 2 us- ing cluster-based Monte Carlo, Physical Review Materi- als5, 024604 (2021)
work page 2021
-
[23]
V. L. Vinograd, E. A. Juarez-Arellano, A. Lieb, K. Knorr, W. Schnick, J. D. Gale, and B. Winkler, Coupled Al/Si and O/N order/disorder in BaYb [Si 4−xAlxOxN7−x] SiAlON, Zeitschrift f¨ ur Kristallographie , 402 (2007)
work page 2007
-
[24]
I. Batatia, D. P. Kovacs, G. Simm, C. Ortner, and G. Cs´ anyi, MACE: Higher order equivariant message passing neural networks for fast and accurate force fields, Advances in Neural Information Processing Systems35, 11423 (2022)
work page 2022
-
[25]
I. Batatia, S. Batzner, D. P. Kov´ acs, A. Musaelian, G. N. Simm, R. Drautz, C. Ortner, B. Kozinsky, and G. Cs´ anyi, The design space of e (3)-equivariant atom-centred in- teratomic potentials, Nature Machine Intelligence7, 56 (2025)
work page 2025
-
[26]
M. Lax, The Franck-Condon principle and its application to crystals, The Journal of chemical physics20, 1752 (1952)
work page 1952
-
[27]
R. Kubo and Y. Toyozawa, Application of the method of generating function to radiative and non-radiative tran- sitions of a trapped electron in a crystal, Progress of The- oretical Physics13, 160 (1955)
work page 1955
-
[28]
J. Bouquiaux, S. Ponc´ e, Y. Jia, A. Miglio, M. Mikami, and X. Gonze, A First-Principles Explanation of the Lu- 10 minescent Line Shape of SrLiAl 3N4: Eu 2+ Phosphor for Light-Emitting Diode Applications, Chemistry of Mate- rials35, 5353 (2023)
work page 2023
-
[29]
M. E. Turiansky and J. L. Lyons, Approximate excited- state potential energy surfaces for defects in solids, Phys- ical Review B112, 205401 (2025)
work page 2025
-
[30]
X. Gonze, J.-M. Beuken, R. Caracas, F. Detraux, M. Fuchs, G.-M. Rignanese, L. Sindic, M. Verstraete, G. Zerah, F. Jollet,et al., First-principles computation of material properties: the ABINIT software project, Com- putational Materials Science25, 478 (2002)
work page 2002
-
[31]
M. J. Verstraete, J. Abreu, G. E. Allemand, B. Amadon, G. Antonius, M. Azizi, L. Baguet, C. Barat, L. Bas- togne, R. B´ ejaud,et al., Abinit 2025: New capabilities for the predictive modeling of solids and nanomaterials, The Journal of Chemical Physics163(2025)
work page 2025
-
[32]
M. Torrent, F. Jollet, F. Bottin, G. Z´ erah, and X. Gonze, Implementation of the projector augmented- wave method in the ABINIT code: Application to the study of iron under pressure, Comput. Mater. Sci.42, 337 (2008)
work page 2008
-
[33]
J. P. Perdew, K. Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77, 3865 (1996)
work page 1996
- [34]
-
[35]
V. Anisimov and O. Gunnarsson, Density-functional cal- culation of effective Coulomb interactions in metals, Physical Review B43, 7570 (1991)
work page 1991
-
[36]
X. Gonze and C. Lee, Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation the- ory, Physical Review B55, 10355 (1997)
work page 1997
-
[37]
A. Togo and I. Tanaka, First principles phonon calcu- lations in materials science, Scripta Materialia108, 1 (2015)
work page 2015
-
[38]
A. Togo, First-principles phonon calculations with phonopy and phono3py, Journal of the Physical Society of Japan92, 012001 (2023)
work page 2023
-
[39]
L. Razinkovas, M. W. Doherty, N. B. Manson, C. G. Van de Walle, and A. Alkauskas, Vibrational and vi- bronic structure of isolated point defects: The nitrogen- vacancy center in diamond, Physical Review B104, 045303 (2021)
work page 2021
-
[40]
J. Bouquiaux, M. Giantomassi, S. Ponc´ e, Y. Jia, M. Mikami, and X. Gonze, Lumabi: a python package to streamline the computation of phonon-resolved lumi- nescence spectra of defects and dopants in solids (2026)
work page 2026
- [42]
-
[43]
A. Loew, D. Sun, H.-C. Wang, S. Botti, and M. A. Mar- ques, Universal machine learning interatomic potentials are ready for phonons, npj Computational Materials11, 178 (2025)
work page 2025
-
[44]
J. Zhou, X. Li, M. Huang, and S. Chen, One-defect one- potential strategy for accurate machine learning predic- tion of phonons in defect-containing supercells, Physical Review B112, 235205 (2025)
work page 2025
-
[45]
M. E. Turiansky, J. L. Lyons, and N. Bernstein, Machine learning phonon spectra for fast and accurate optical line- shapes of defects, ACS nano (2025)
work page 2025
-
[46]
J. Bouquiaux, M. Giantomassi, S. Ponc´ e, Y. Jia, M. Mikami, and X. Gonze, Lumabi: a python package to streamline the computation of phonon-resolved lumi- nescence spectra of defects and dopants in solids, Journal of Open Source Software11, 9145 (2026)
work page 2026
-
[47]
P. B. Allen, T. Berlijn, D. Casavant, and J. Soler, Re- covering hidden Bloch character: Unfolding electrons, phonons, and slabs, Physical Review B—Condensed Matter and Materials Physics87, 085322 (2013)
work page 2013
-
[48]
V. Popescu and A. Zunger, Extracting E versus k effec- tive band structure from supercell calculations on alloys and impurities, Physical Review B—Condensed Matter and Materials Physics85, 085201 (2012)
work page 2012
-
[49]
A. Togo, L. Chaput, T. Tadano, and I. Tanaka, Imple- mentation strategies in phonopy and phono3py, Journal of Physics: Condensed Matter35, 353001 (2023). SI: Micro-environment of the Eu interstitial in theβ-SiAlON:Eu 2+ green phosphor Julien Bouquiaux,1, 2,∗ Samuel Ponc´ e,1, 3 Yongchao Jia,4 Masayoshi Mikami,5 and Xavier Gonze 1 1Institute of Condensed M...
work page 2023
-
[50]
plane than Eu, and six longer ones (d≈2.74– 2.84 ˚A). Table S3 gives the second-shell composition (number of Al, N, O, and Si atoms within∼5 ˚A of Eu). Figure S8 shows the computed PL lineshapes atT= 6 K for all 14 configurations, grouped by composition. Calculations are done in the 225-atoms supercell and with machine-learnedMatterSimphonons. Within each...
work page 2000
-
[51]
Ilyes Batatia, David P Kovacs, Gregor Simm, Christoph Ortner, and G´ abor Cs´ anyi. MACE: Higher order equiv- ariant message passing neural networks for fast and ac- curate force fields.Advances in Neural Information Pro- cessing Systems, 35:11423–11436, 2022
work page 2022
-
[52]
Ilyes Batatia, Simon Batzner, D´ avid P´ eter Kov´ acs, Albert Musaelian, Gregor NC Simm, Ralf Drautz, Christoph Ortner, Boris Kozinsky, and G´ abor Cs´ anyi. The design space of e (3)-equivariant atom-centred inter- atomic potentials.Nature Machine Intelligence, 7(1):56– 67, 2025
work page 2025
-
[53]
Zhenbin Wang, Weike Ye, Iek-Heng Chu, and Shyue Ping Ong. Elucidating structure–composition–property rela- tionships of theβ-SiAlON: Eu 2+ phosphor.Chemistry of Materials, 28(23):8622–8630, 2016
work page 2016
-
[54]
Audrius Alkauskas, Bob B Buckley, David D Awschalom, and Chris G Van de Walle. First-principles theory of the luminescence lineshape for the triplet transition in diamond NV centres.New J. Phys., 16(7):073026, 2014
work page 2014
-
[55]
Yu Jin, Marco Govoni, Gary Wolfowicz, Sean E Sulli- van, F Joseph Heremans, David D Awschalom, and Giu- lia Galli. Photoluminescence spectra of point defects in semiconductors: Validation of first-principles calcu- lations.Physical Review Materials, 5(8):084603, 2021
work page 2021
-
[56]
Lukas Razinkovas, Marcus W Doherty, Neil B Manson, Chris G Van de Walle, and Audrius Alkauskas. Vibra- tional and vibronic structure of isolated point defects: The nitrogen-vacancy center in diamond.Physical Re- view B, 104(4):045303, 2021
work page 2021
-
[57]
Julien Bouquiaux, Matteo Giantomassi, Samuel Ponc´ e, Yongchao Jia, Masayoshi Mikami, and Xavier Gonze. Lumabi: a python package to streamline the computa- tion of phonon-resolved luminescence spectra of defects and dopants in solids.Journal of Open Source Software, 11(121):9145, 2026
work page 2026
-
[58]
Julien Bouquiaux, Samuel Ponc´ e, Yongchao Jia, Anna Miglio, Masayoshi Mikami, and Xavier Gonze. A First- Principles Explanation of the Luminescent Line Shape of SrLiAl3N4: Eu 2+ Phosphor for Light-Emitting Diode Applications.Chemistry of Materials, 35(14):5353–5361, 2023
work page 2023
-
[59]
arXiv preprint arXiv:2405.04967 , year=
Han Yang, Chenxi Hu, Yichi Zhou, Xixian Liu, Yu Shi, Jielan Li, Guanzhi Li, Zekun Chen, Shuizhou Chen, Claudio Zeni, et al. Mattersim: A deep learning atom- istic model across elements, temperatures and pressures. arXiv preprint arXiv:2405.04967, 2024
-
[60]
Xinyu Zhang, Wanyuan Li, Chao Wang, Lin Zhang, Shuxing Li, and Rong-Jun Xie. Narrow-band green ni- tride phosphor for wide color gamut backlighting.Chem- ical Engineering Journal, 509:161073, 2025
work page 2025
-
[61]
First-principles study of the luminescence of Eu 2+-doped phosphors.Phys
Yongchao Jia, Anna Miglio, Samuel Ponc´ e, Masayoshi Mikami, and Xavier Gonze. First-principles study of the luminescence of Eu 2+-doped phosphors.Phys. Rev. B, 96(12):125132, 2017
work page 2017
-
[62]
Julien Bouquiaux, Samuel Ponc´ e, Yongchao Jia, Anna Miglio, Masayoshi Mikami, and Xavier Gonze. Impor- tance of Long-Range Channel Sr Displacements for the Narrow Emission in Sr[Li2Al2O2N2]:Eu2+ Phosphor.Ad- vanced Optical Materials, 9(20):2100649, 2021
work page 2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.