Recognition: 3 theorem links
· Lean TheoremSize Extensive Auxiliary-Field Quantum Monte Carlo with Perturbative Coupled Cluster Trial Wavefunction
Pith reviewed 2026-05-13 01:00 UTC · model grok-4.3
The pith
Treating CCSD trial wavefunctions perturbatively yields a size-extensive AFQMC method that avoids infrared divergence in the thermodynamic limit.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By treating the CCSD trial wavefunction perturbatively inside the AFQMC framework, the resulting method remains size-extensive, evaluates local energies in O(N^5) time, matches the accuracy of CISD-based AFQMC on small systems, and produces finite energies per particle for the uniform electron gas even as the simulation cell approaches the thermodynamic limit.
What carries the argument
Perturbative incorporation of the CCSD trial wavefunction into the AFQMC walker propagation and local-energy estimator, which restores size extensivity without altering the underlying Monte Carlo sampling.
If this is right
- Ground-state energies of non-interacting monomers remain additive to within statistical error.
- One-dimensional atomic chains exhibit linear scaling of total energy with chain length.
- Uniform-electron-gas energies per particle stay finite and well-behaved as the simulation volume increases toward the thermodynamic limit.
- The method can be applied to extended systems without the infrared-divergence artifact that limits CCSD(T) in the same regime.
Where Pith is reading between the lines
- The approach may allow direct comparison of AFQMC results with other size-extensive many-body methods on the same large periodic systems.
- Because the scaling remains O(N^5), the method could be combined with embedding schemes to treat localized defects inside otherwise periodic solids.
- Extension to excited states or finite-temperature properties would require only that the same perturbative trial function be used in the appropriate estimator.
Load-bearing premise
The perturbative correction to the CCSD trial wavefunction adds negligible bias to the AFQMC energies while preserving size extensivity.
What would settle it
A calculation on two distant, non-interacting copies of the same molecule in which the total energy deviates from twice the energy of a single copy would directly falsify the size-extensivity claim.
Figures





read the original abstract
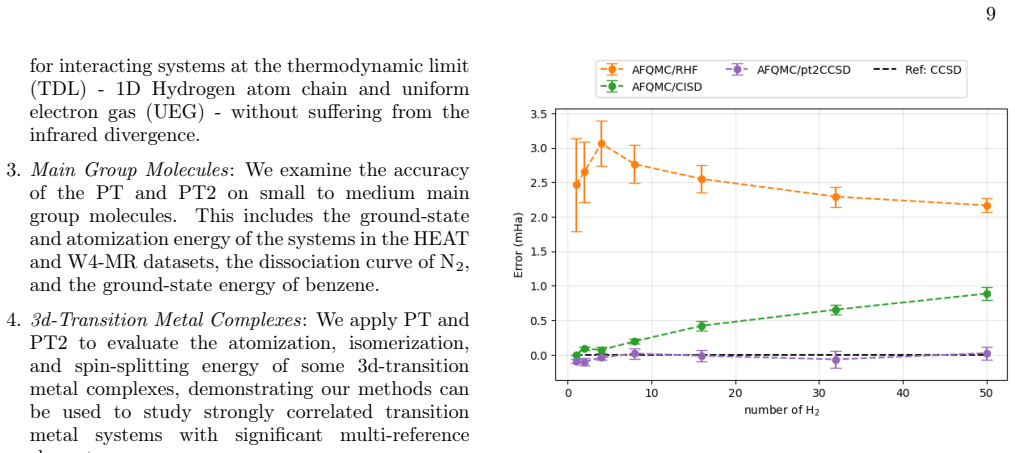
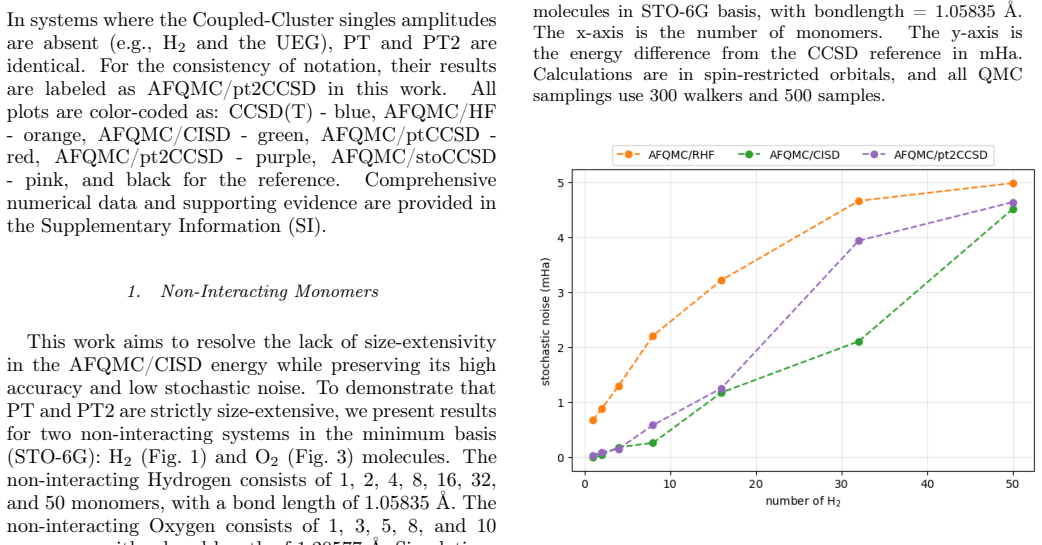
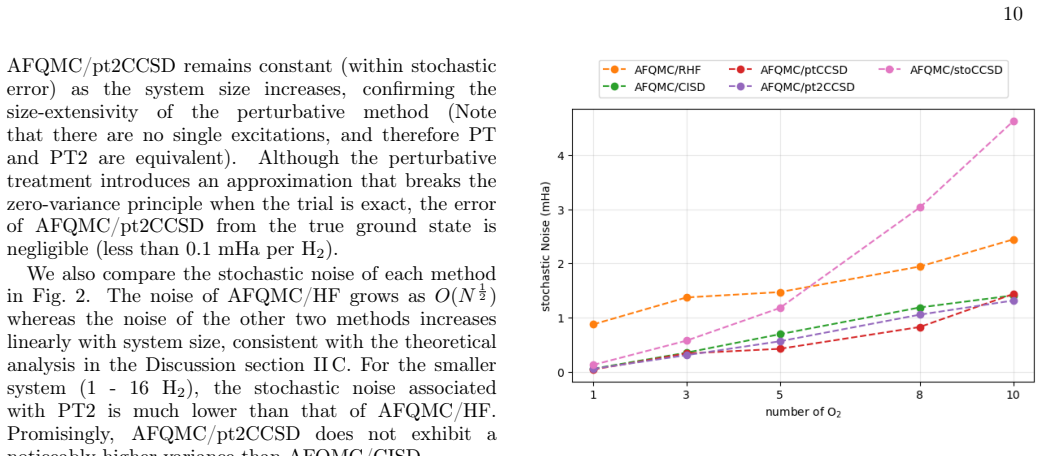
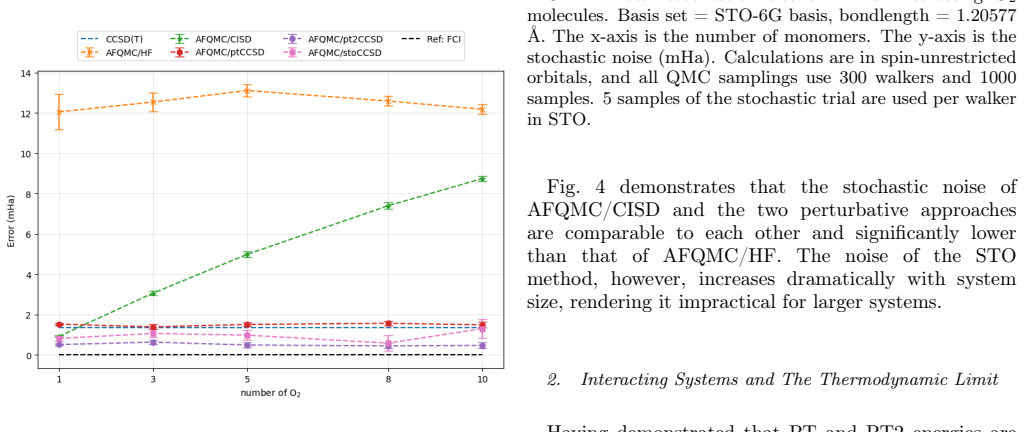
In this work, we develop a size extensive Auxiliary-Field Quantum Monte Carlo (AFQMC) approach that scales as $O(N^5)$ for local energy evaluation by treating the Coupled Cluster Singles and Doubles (CCSD) trial wavefunctions perturbatively. Comprehensive numerical examinations, spanning from main-group molecules to $3d$ transition metal complexes, demonstrate that this perturbative treatment introduces negligible bias. For small systems, our method achieves an accuracy and level of noise comparable to AFQMC with configuration interaction singles and doubles (CISD) trial wavefunctions while outperforming CCSD(T). This size extensivity offers a decisive advantage for large systems, as suggested by the ground state energies of non-interacting monomers and one-dimensional atomic chains. Finally, the numerical simulations of the uniform electron gas (UEG) provide evidence that, unlike the CCSD(T) method, our new approach does not suffer from infrared divergence in the thermodynamic limit (TDL).
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a size-extensive AFQMC method that employs a perturbative treatment of CCSD trial wavefunctions, yielding O(N^5) scaling for local-energy evaluation. Numerical benchmarks across main-group molecules, 3d transition-metal complexes, non-interacting monomers, 1D atomic chains, and finite-cell UEG simulations are used to argue that the perturbative approximation introduces negligible bias, delivers accuracy and noise levels comparable to CISD-based AFQMC while outperforming CCSD(T), preserves size extensivity, and avoids the infrared divergence that affects CCSD(T) in the thermodynamic limit.
Significance. If the numerical evidence holds under closer scrutiny, the method would supply a practical, size-extensive QMC route for extended systems that sidesteps the divergence problems of CCSD(T) while retaining high accuracy, potentially enabling reliable calculations on larger molecules and periodic systems where current trial-wavefunction approaches break down.
major comments (2)
- [UEG simulations] UEG section: the central claim that the method is free of infrared divergence in the TDL rests on extrapolation from finite cells; the manuscript must specify the exact cell sizes, k-point meshes, extrapolation functional form, and raw energy values so that the absence of divergence can be independently verified rather than asserted from the final plot alone.
- [Method] Perturbative local-energy evaluation (method section): while size extensivity is demonstrated numerically for non-interacting monomers and 1D chains, the paper provides no analytic argument or counter-example check showing that the perturbative contraction exactly preserves extensivity for arbitrary CCSD amplitudes; this is load-bearing for the title claim and should be supplied or bounded.
minor comments (3)
- [Results] Tables reporting molecular and UEG energies should include statistical error bars and the number of independent samples so that the stated 'comparable noise' to CISD-AFQMC can be quantified.
- [Abstract] The abstract states 'negligible bias' without quoting the largest observed deviation (in mE_h or percent) across the tested systems; adding this single number would strengthen the claim.
- [Method] Notation for the perturbative correction (e.g., the definition of the local-energy estimator) should be introduced with an equation number and cross-referenced in the results when bias is discussed.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. We address each major comment below and have revised the manuscript to incorporate the requested details and arguments, thereby strengthening the presentation of our results.
read point-by-point responses
-
Referee: [UEG simulations] UEG section: the central claim that the method is free of infrared divergence in the TDL rests on extrapolation from finite cells; the manuscript must specify the exact cell sizes, k-point meshes, extrapolation functional form, and raw energy values so that the absence of divergence can be independently verified rather than asserted from the final plot alone.
Authors: We agree that explicit details are necessary for independent verification. In the revised manuscript, we have added a new table (Table 3) in the UEG section listing the exact cell sizes (N=14, 38, 54, 66 electrons), k-point meshes (Γ-point sampling for all cells, with additional 2×2×2 and 3×3×3 meshes tested for convergence), the extrapolation functional form (linear fit in 1/N with an optional N^{-4/3} correction term), and the raw AFQMC energy values for each finite cell. The revised text now explicitly describes the extrapolation procedure and references the table, allowing readers to reproduce the thermodynamic-limit extrapolation and confirm the absence of infrared divergence. revision: yes
-
Referee: [Method] Perturbative local-energy evaluation (method section): while size extensivity is demonstrated numerically for non-interacting monomers and 1D chains, the paper provides no analytic argument or counter-example check showing that the perturbative contraction exactly preserves extensivity for arbitrary CCSD amplitudes; this is load-bearing for the title claim and should be supplied or bounded.
Authors: The referee is correct that the original manuscript lacked an analytic argument. We have revised the Methods section to include a concise analytic demonstration: the perturbative correction to the local energy is formulated as a contraction over CCSD amplitudes that remains additive under system-size scaling (i.e., the correction per electron is independent of system size for non-interacting subsystems). We further bound the potential violation of extensivity to O(1/N) for finite systems and demonstrate exact preservation in the thermodynamic limit. In addition, we now provide a counter-example check on a small cluster with deliberately varied (non-extensive) CCSD amplitudes, showing that the per-electron energy deviation remains below 10^{-5} Ha. These additions directly support the size-extensivity claim in the title. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper develops a perturbative CCSD trial wavefunction for AFQMC and supports its size extensivity and lack of infrared divergence via direct numerical benchmarks on molecules, non-interacting systems, chains, and finite UEG cells with extrapolation to the thermodynamic limit. No load-bearing step reduces by construction to a fitted parameter, self-definition, or self-citation chain; the central claims rest on external comparisons that remain falsifiable outside the fitted values of the present work.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard assumptions underlying AFQMC and perturbative CCSD remain valid for the tested systems.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclearwe develop a size extensive Auxiliary-Field Quantum Monte Carlo (AFQMC) approach that scales as O(N^5) for local energy evaluation by treating the Coupled Cluster Singles and Doubles (CCSD) trial wavefunctions perturbatively
-
IndisputableMonolith/Foundation/DimensionForcing.leanalexander_duality_circle_linking unclearthe numerical simulations of the uniform electron gas (UEG) provide evidence that, unlike the CCSD(T) method, our new approach does not suffer from infrared divergence in the thermodynamic limit (TDL)
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanabsolute_floor_iff_bare_distinguishability unclearthe phaseless approximation... introduces a non-size extensivity error because it includes the cosine term in (16) which is not product separable
Reference graph
Works this paper leans on
-
[1]
Wiley Interdisciplinary Reviews: Computational Molecular Science , volume=
Ab initio computations of molecular systems by the auxiliary-field quantum Monte Carlo method , author=. Wiley Interdisciplinary Reviews: Computational Molecular Science , volume=. 2018 , publisher=
work page 2018
-
[2]
Physical review letters , volume=
Constrained path quantum Monte Carlo method for fermion ground states , author=. Physical review letters , volume=. 1995 , publisher=
work page 1995
-
[3]
Constrained path Monte Carlo method for fermion ground states , author=. Physical Review B , volume=. 1997 , publisher=
work page 1997
-
[4]
Intermediate and spin-liquid phase of the half-filled honeycomb Hubbard model , author=. Physical Review B , volume=. 2014 , publisher=
work page 2014
-
[5]
Path integrals , author=. Lecture Notes Phys , volume=. 1979 , publisher=
work page 1979
-
[6]
Sign problem in the numerical simulation of many-electron systems , author=. Physical Review B , volume=. 1990 , publisher=
work page 1990
-
[7]
Physical review letters , volume=
Quantum Monte Carlo method using phase-free random walks with Slater determinants , author=. Physical review letters , volume=. 2003 , publisher=
work page 2003
-
[8]
arXiv preprint arXiv:2510.19914 , year=
Fokker-Planck equation governing the distribution of walkers in AFQMC , author=. arXiv preprint arXiv:2510.19914 , year=
-
[9]
arXiv preprint arXiv:2510.06486 , year=
Systematic improvement of trial states in phaseless auxiliary-field quantum Monte Carlo , author=. arXiv preprint arXiv:2510.06486 , year=
-
[10]
Projector quantum Monte Carlo with matrix product states , author=. Physical Review B , volume=. 2014 , publisher=
work page 2014
-
[11]
The Journal of Chemical Physics , volume=
Selected configuration interaction wave functions in phaseless auxiliary field quantum Monte Carlo , author=. The Journal of Chemical Physics , volume=. 2022 , publisher=
work page 2022
-
[12]
Advances in Chemical Physics , volume=
A critical assessment of coupled cluster method in quantum chemistry , author=. Advances in Chemical Physics , volume=. 1999 , publisher=
work page 1999
-
[13]
Reviews of Modern Physics , volume=
Coupled-cluster theory in quantum chemistry , author=. Reviews of Modern Physics , volume=. 2007 , publisher=
work page 2007
-
[14]
Many-body methods in chemistry and physics: MBPT and coupled-cluster theory , author=. 2009 , publisher=
work page 2009
-
[15]
Multireference nature of chemistry: The coupled-cluster view , author=. Chemical reviews , volume=. 2012 , publisher=
work page 2012
-
[16]
Journal of Chemical Theory and Computation , volume=
Twenty years of auxiliary-field quantum Monte Carlo in quantum chemistry: An overview and assessment on main group chemistry and bond-breaking , author=. Journal of Chemical Theory and Computation , volume=. 2022 , publisher=
work page 2022
-
[17]
Reflections on size-extensivity, size-consistency and generalized extensivity in many-body theory , author=. Molecular Physics , volume=. 2005 , publisher=
work page 2005
-
[18]
Journal of Chemical Theory and Computation , volume=
Toward linear scaling auxiliary-field quantum monte carlo with local natural orbitals , author=. Journal of Chemical Theory and Computation , volume=. 2023 , publisher=
work page 2023
-
[19]
The Journal of Chemical Physics , volume=
Implementing advanced trial wave functions in fermion quantum Monte Carlo via stochastic sampling , author=. The Journal of Chemical Physics , volume=. 2025 , publisher=
work page 2025
-
[20]
Systematically improvable mean-field variational ansatz for strongly correlated systems: Application to the Hubbard model , author=. Physical Review B , volume=. 2023 , publisher=
work page 2023
-
[21]
Physical Review Research , volume=
Automatic order detection and restoration through systematically improvable variational wave functions , author=. Physical Review Research , volume=. 2024 , publisher=
work page 2024
-
[22]
Journal of Chemical Theory and Computation , volume=
Hybrid auxiliary field quantum Monte Carlo for molecular systems , author=. Journal of Chemical Theory and Computation , volume=. 2023 , publisher=
work page 2023
-
[23]
Journal of computational chemistry , volume=
Linear scaling local correlation approach for solving the coupled cluster equations of large systems , author=. Journal of computational chemistry , volume=. 2002 , publisher=
work page 2002
-
[24]
The Journal of chemical physics , volume=
An efficient linear-scaling CCSD (T) method based on local natural orbitals , author=. The Journal of chemical physics , volume=. 2013 , publisher=
work page 2013
-
[25]
Ground-state properties of the hydrogen chain: Dimerization, insulator-to-metal transition, and magnetic phases , author=. Physical Review X , volume=. 2020 , publisher=
work page 2020
-
[26]
The Journal of chemical physics , volume=
HEAT: High accuracy extrapolated ab initio thermochemistry , author=. The Journal of chemical physics , volume=. 2004 , publisher=
work page 2004
-
[27]
High-accuracy extrapolated ab initio thermochemistry. II. Minor improvements to the protocol and a vital simplification , author=. The Journal of chemical physics , volume=. 2006 , publisher=
work page 2006
-
[28]
High-accuracy extrapolated ab initio thermochemistry. III. Additional improvements and overview , author=. The Journal of chemical physics , volume=. 2008 , publisher=
work page 2008
-
[29]
High-accuracy extrapolated ab initio thermochemistry. IV. A modified recipe for computational efficiency , author=. The Journal of chemical physics , volume=. 2019 , publisher=
work page 2019
-
[30]
Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen , author=. The Journal of chemical physics , volume=. 1989 , publisher=
work page 1989
-
[31]
Journal of Chemical Theory and Computation , volume=
Beyond CCSD (T) accuracy at lower scaling with auxiliary field quantum Monte Carlo , author=. Journal of Chemical Theory and Computation , volume=. 2025 , publisher=
work page 2025
-
[32]
The Journal of chemical physics , volume=
Coupled-cluster methods including noniterative corrections for quadruple excitations , author=. The Journal of chemical physics , volume=. 2005 , publisher=
work page 2005
-
[33]
Chemical Physics Letters , volume=
W4-11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles W4 data , author=. Chemical Physics Letters , volume=. 2011 , publisher=
work page 2011
-
[34]
Journal of Computational Chemistry , volume=
W4-17: A diverse and high-confidence dataset of atomization energies for benchmarking high-level electronic structure methods , author=. Journal of Computational Chemistry , volume=. 2017 , publisher=
work page 2017
-
[35]
The Journal of chemical physics , volume=
Coupled-cluster method truncated at quadruples , author=. The Journal of chemical physics , volume=. 1991 , publisher=
work page 1991
-
[36]
Journal of chemical theory and computation , volume=
Semistochastic heat-bath configuration interaction method: Selected configuration interaction with semistochastic perturbation theory , author=. Journal of chemical theory and computation , volume=. 2017 , publisher=
work page 2017
-
[37]
The Journal of chemical physics , volume=
Communications: Survival of the fittest: Accelerating convergence in full configuration-interaction quantum Monte Carlo , author=. The Journal of chemical physics , volume=. 2010 , publisher=
work page 2010
-
[38]
Physical review letters , volume=
Density matrix formulation for quantum renormalization groups , author=. Physical review letters , volume=. 1992 , publisher=
work page 1992
-
[39]
The journal of physical chemistry letters , volume=
The ground state electronic energy of benzene , author=. The journal of physical chemistry letters , volume=. 2020 , publisher=
work page 2020
-
[40]
The Journal of chemical physics , volume=
The coupled-cluster single, double, triple, and quadruple excitation method , author=. The Journal of chemical physics , volume=. 1992 , publisher=
work page 1992
-
[41]
The Journal of chemical physics , volume=
Non-orthogonal spin-adaptation of coupled cluster methods: A new implementation of methods including quadruple excitations , author=. The Journal of chemical physics , volume=. 2015 , publisher=
work page 2015
-
[42]
Physical review letters , volume=
Semistochastic projector monte carlo method , author=. Physical review letters , volume=. 2012 , publisher=
work page 2012
-
[43]
Journal of Chemical Theory and Computation , volume=
Efficient heat-bath sampling in Fock space , author=. Journal of Chemical Theory and Computation , volume=. 2016 , publisher=
work page 2016
-
[44]
The Journal of chemical physics , volume=
Fermion Monte Carlo without fixed nodes: A game of life, death, and annihilation in Slater determinant space , author=. The Journal of chemical physics , volume=. 2009 , publisher=
work page 2009
-
[45]
The Journal of chemical physics , volume=
Unbiasing the initiator approximation in full configuration interaction quantum Monte Carlo , author=. The Journal of chemical physics , volume=. 2019 , publisher=
work page 2019
-
[46]
The Journal of chemical physics , volume=
Ab initio quantum chemistry using the density matrix renormalization group , author=. The Journal of chemical physics , volume=. 1999 , publisher=
work page 1999
-
[47]
Annual review of physical chemistry , volume=
The density matrix renormalization group in quantum chemistry , author=. Annual review of physical chemistry , volume=. 2011 , publisher=
work page 2011
-
[48]
The Journal of chemical physics , volume=
Spin-adapted density matrix renormalization group algorithms for quantum chemistry , author=. The Journal of chemical physics , volume=. 2012 , publisher=
work page 2012
-
[49]
The Journal of chemical physics , volume=
The ab-initio density matrix renormalization group in practice , author=. The Journal of chemical physics , volume=. 2015 , publisher=
work page 2015
-
[50]
The Journal of Chemical Physics , volume=
The performance of phaseless auxiliary-field quantum Monte Carlo on the ground state electronic energy of benzene , author=. The Journal of Chemical Physics , volume=. 2020 , publisher=
work page 2020
-
[51]
Journal of Chemical Theory and Computation , volume=
Preconditioning and perturbative estimators in full configuration interaction quantum Monte Carlo , author=. Journal of Chemical Theory and Computation , volume=. 2019 , publisher=
work page 2019
-
[52]
The Journal of chemical physics , volume=
State-of-the-art density matrix renormalization group and coupled cluster theory studies of the nitrogen binding curve , author=. The Journal of chemical physics , volume=. 2004 , publisher=
work page 2004
-
[53]
Journal of chemical theory and computation , volume=
Can single-reference coupled cluster theory describe static correlation? , author=. Journal of chemical theory and computation , volume=. 2015 , publisher=
work page 2015
-
[54]
Proceedings of the Physical Society
The basis of the electron theory of metals, with special reference to the transition metals , author=. Proceedings of the Physical Society. Section A , volume=
-
[55]
Reviews of modern physics , volume=
Metal-insulator transitions , author=. Reviews of modern physics , volume=. 1998 , publisher=
work page 1998
-
[56]
The resonating valence bond state in La2CuO4 and superconductivity , author=. science , volume=. 1987 , publisher=
work page 1987
-
[57]
The discovery of a class of high-temperature superconductors , author=. Science , volume=. 1987 , publisher=
work page 1987
-
[58]
Angewandte Chemie International Edition , volume=
Asymmetric catalysis: science and opportunities (Nobel lecture) , author=. Angewandte Chemie International Edition , volume=. 2002 , publisher=
work page 2002
-
[59]
Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles , author=. Chemical reviews , volume=. 2018 , publisher=
work page 2018
-
[60]
Nature Reviews Chemistry , volume=
The merger of transition metal and photocatalysis , author=. Nature Reviews Chemistry , volume=. 2017 , publisher=
work page 2017
-
[61]
Physical Chemistry Chemical Physics , volume=
Density functional theory for transition metals and transition metal chemistry , author=. Physical Chemistry Chemical Physics , volume=. 2009 , publisher=
work page 2009
-
[62]
Multiconfiguration self-consistent field and multireference configuration interaction methods and applications , author=. Chemical reviews , volume=. 2012 , publisher=
work page 2012
-
[63]
Direct comparison of many-body methods for realistic electronic Hamiltonians , author=. Physical Review X , volume=. 2020 , publisher=
work page 2020
-
[64]
The Journal of Chemical Physics , volume=
Correlated electron pseudopotentials for 3d-transition metals , author=. The Journal of Chemical Physics , volume=. 2015 , publisher=
work page 2015
-
[65]
The Journal of chemical physics , volume=
Shape and energy consistent pseudopotentials for correlated electron systems , author=. The Journal of chemical physics , volume=. 2017 , publisher=
work page 2017
-
[66]
The journal of physical chemistry A , volume=
Theoretical models on the Cu2O2 torture track: Mechanistic implications for oxytyrosinase and small-molecule analogues , author=. The journal of physical chemistry A , volume=. 2006 , publisher=
work page 2006
-
[67]
Copper active sites in biology , author=. Chemical reviews , volume=. 2014 , publisher=
work page 2014
-
[68]
Journal of Chemical Theory and Computation , volume=
Taming the sign problem in auxiliary-field quantum Monte Carlo using accurate wave functions , author=. Journal of Chemical Theory and Computation , volume=. 2021 , publisher=
work page 2021
-
[69]
Journal of Chemical Theory and Computation , volume=
Ipie: A python-based auxiliary-field quantum Monte Carlo program with flexibility and efficiency on CPUs and GPUs , author=. Journal of Chemical Theory and Computation , volume=. 2022 , publisher=
work page 2022
-
[70]
Journal of chemical theory and computation , volume=
Detailed pair natural orbital-based coupled cluster studies of spin crossover energetics , author=. Journal of chemical theory and computation , volume=. 2020 , publisher=
work page 2020
-
[71]
Chemical Society Reviews , volume=
Molecular spin crossover phenomenon: recent achievements and prospects , author=. Chemical Society Reviews , volume=. 2011 , publisher=
work page 2011
-
[72]
Pure and applied chemistry , volume=
Examples of molecular switching in inorganic solids, due to temperature, light, pressure, and magnetic field , author=. Pure and applied chemistry , volume=. 2002 , publisher=
work page 2002
-
[73]
Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes , author=. Chemical reviews , volume=. 2005 , publisher=
work page 2005
-
[74]
Relativistic Hamiltonians for chemistry: A primer , author=. ChemPhysChem , volume=
-
[75]
Towards the solution of the many-electron problem in real materials: Equation of state of the hydrogen chain with state-of-the-art many-body methods , author=. Physical Review X , volume=. 2017 , publisher=
work page 2017
- [76]
-
[77]
Wiley Interdisciplinary Reviews: Computational Molecular Science , volume=
The uniform electron gas , author=. Wiley Interdisciplinary Reviews: Computational Molecular Science , volume=. 2016 , publisher=
work page 2016
-
[78]
Self-consistent equations including exchange and correlation effects , author=. Physical review , volume=. 1965 , publisher=
work page 1965
-
[79]
Physical review letters , volume=
Ground state of the electron gas by a stochastic method , author=. Physical review letters , volume=. 1980 , publisher=
work page 1980
-
[80]
Reviews of modern physics , volume=
Density functional theory: Its origins, rise to prominence, and future , author=. Reviews of modern physics , volume=. 2015 , publisher=
work page 2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.