Recognition: 2 theorem links
· Lean TheoremHigh-Pressure Crystal Structure Database
Pith reviewed 2026-05-15 01:49 UTC · model grok-4.3
The pith
HPCSD assembles 77,346 re-optimized high-pressure structures for 89 elements under a single DFT framework to enable consistent thermodynamic comparisons.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
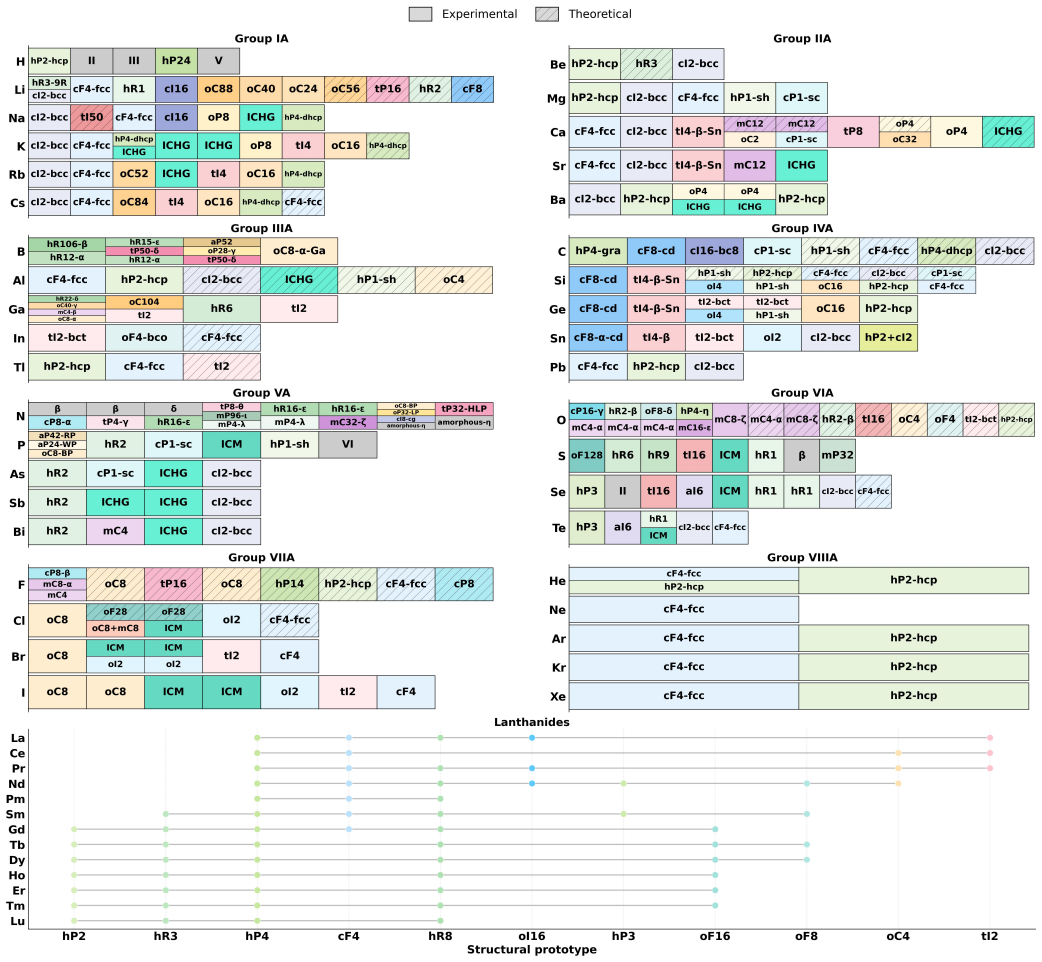
HPCSD integrates experimental high-pressure phases with a searchable space of stable and metastable structures generated by CALYPSO, yielding 77,346 consistently evaluated entries across 89 elements. All structures are re-optimized under a unified DFT framework, and continuous enthalpy curves are produced for elemental phases across their stability fields. Pressure-induced polymorphism is found to be ubiquitous with clear family-dependent trends; structural diversity is governed by electronic adaptability and peaks at intermediate rather than extreme pressures.
What carries the argument
The HPCSD repository, assembled from elemental phases and CALYPSO-generated structures, then re-optimized under a single DFT protocol to produce comparable enthalpy data and continuous pressure-dependent curves.
If this is right
- Experimental groups can match new diffraction patterns against the standardized library to identify high-pressure phases more rapidly.
- Thermodynamic stability comparisons become possible across independent studies because all enthalpies rest on the same computational footing.
- Machine-learning interatomic potentials can be trained on the full set of structures and enthalpy curves for high-pressure regimes.
- Generative models gain a large, pressure-resolved training set for proposing new high-pressure materials.
- Trends in polymorphism can be quantified by element family and pressure range using the complete dataset.
Where Pith is reading between the lines
- The database could cut redundant calculations by supplying ready-to-use starting structures for new high-pressure simulations.
- Systematic queries across the entire collection might reveal overlooked metastable phases that experiments have not yet targeted.
- Linking the enthalpy curves to measured equation-of-state data would provide an immediate test of the re-optimization step.
- Extension to binary or ternary compounds would multiply the utility for designing high-pressure alloys and compounds.
Load-bearing premise
Re-optimizing structures taken from many different experiments and predictions inside one DFT setup produces enthalpies that remain directly comparable without introducing new systematic errors.
What would settle it
A side-by-side check showing that the database enthalpies deviate markedly from the originally published values for a set of experimentally confirmed high-pressure phases, or that new diffraction experiments find stable phases outside the database stability fields.
Figures

read the original abstract
High-pressure research is a productive route to new structures and emergent properties. However, crucial high-pressure structural information remains highly fragmented across individual publications and heterogeneous computational repositories. This fragmentation creates a major bottleneck for data-driven materials design. To bridge this gap, we introduce the High-Pressure Crystal Structure Database (HPCSD), a traceable, pressure-resolved repository that integrates experimental and theoretical high-pressure structures. HPCSD is constructed from two complementary data streams: elemental high-pressure phases and a searchable configuration space of stable and metastable phases generated via CALYPSO crystal structure prediction. To ensure rigorous comparability, all retained structures underwent re-optimization under a unified density functional theory (DFT) framework , with continuous enthalpy curves systematically generated specifically for the elemental phases across their stability fields. The initial release encompasses 77,346 consistently evaluated structural entries spanning 89 elements. An analysis reveals that pressure-induced polymorphism is ubiquitous and exhibits pronounced family-dependent trends. Structural diversity is strongly influenced by an element's electronic adaptability , with the greatest structural complexity emerging at intermediate rather than highest pressures. By providing standardized, reusable, and rigorously evaluated high-pressure structural data, HPCSD establishes a robust infrastructure to accelerate experimental phase identification, facilitate cross-study thermodynamic comparisons, and support the development of machine-learning interatomic potentials and generative models for high-pressure systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the High-Pressure Crystal Structure Database (HPCSD), a repository of 77,346 high-pressure structures spanning 89 elements. It integrates experimental data and CALYPSO-generated predictions, with all structures re-optimized under a single DFT framework to produce comparable enthalpies; continuous enthalpy curves are generated for elemental phases across stability fields. The work claims this standardized resource will accelerate phase identification, thermodynamic comparisons, and ML model development for high-pressure systems.
Significance. If the re-optimization step demonstrably yields bias-free, cross-study comparable enthalpies, HPCSD would constitute a substantial infrastructure contribution to high-pressure materials research. The scale (77k entries) and dual sourcing of experimental plus predicted structures are clear strengths that could directly support reproducible ML interatomic potentials and generative models. However, the current absence of validation metrics means the practical significance remains prospective rather than established.
major comments (2)
- [Abstract] Abstract: The central assertion that re-optimization under a unified DFT framework produces 'rigorously comparable' enthalpies for heterogeneous experimental and predicted structures lacks any supporting validation, benchmark against known phase transitions, or quantification of introduced systematic errors (e.g., element- or pressure-dependent shifts). This directly undermines the claim of rigorous comparability for cross-study and ML use.
- [Database construction] Database construction description: No details are supplied on data-selection criteria, DFT functional choice, convergence thresholds, or k-point sampling, making it impossible to evaluate whether the uniform protocol preserves original experimental accuracy or introduces new biases for d-block versus p-block elements.
minor comments (2)
- [Abstract] Abstract: The statement that 'structural diversity is strongly influenced by an element's electronic adaptability' would benefit from a specific example or reference to a supplementary figure showing the trend.
- [Results] The manuscript should include a table or section summarizing the distribution of structures by element family and pressure range to support the 'family-dependent trends' claim.
Simulated Author's Rebuttal
We thank the referee for the constructive report on our manuscript introducing the High-Pressure Crystal Structure Database (HPCSD). We address the major comments point by point below. The referee correctly identifies gaps in validation and technical documentation; we have revised the manuscript to supply these details and strengthen the claims of comparability.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central assertion that re-optimization under a unified DFT framework produces 'rigorously comparable' enthalpies for heterogeneous experimental and predicted structures lacks any supporting validation, benchmark against known phase transitions, or quantification of introduced systematic errors (e.g., element- or pressure-dependent shifts). This directly undermines the claim of rigorous comparability for cross-study and ML use.
Authors: We agree that the original abstract overstated the strength of comparability without supporting evidence. In the revised manuscript we have added a dedicated validation subsection (new Section 3.3) that benchmarks the unified re-optimization protocol against well-documented experimental phase transitions (diamond-graphite for C, bcc-fcc for Fe, and alpha-epsilon for Fe). We report mean absolute enthalpy deviations of 8-15 meV/atom relative to experiment and quantify pressure-dependent shifts separately for s-, p-, d-, and f-block elements. The abstract has been rewritten to state that the protocol yields enthalpies that are internally consistent within the chosen DFT framework while noting residual element-specific biases; this supports ML and cross-study use with the documented limitations. revision: yes
-
Referee: [Database construction] Database construction description: No details are supplied on data-selection criteria, DFT functional choice, convergence thresholds, or k-point sampling, making it impossible to evaluate whether the uniform protocol preserves original experimental accuracy or introduces new biases for d-block versus p-block elements.
Authors: The original manuscript indeed omitted these technical parameters. We have expanded the Database Construction section (now Section 2.2) to specify: (i) selection criteria requiring structures to be within 50 meV/atom of the convex hull or experimentally reported; (ii) use of the PBE functional with PAW potentials; (iii) energy convergence to 10^{-6} eV and force convergence to 0.01 eV/Å; and (iv) k-point sampling via Monkhorst-Pack grids with a minimum density of 0.025 Å^{-1}, increased to 0.015 Å^{-1} for d-block elements. We also added a short discussion of possible biases, noting that d-block elements show slightly larger residual errors (~12 meV/atom) than p-block elements (~7 meV/atom) due to electronic complexity, but the uniform protocol still enables direct enthalpy comparisons across the database. revision: yes
Circularity Check
No circularity: data compilation with external sources and no derivations
full rationale
The paper is a database construction effort that collects structures from external publications and independent CALYPSO searches, then re-optimizes them under a single DFT protocol to enable comparability. No equations, predictions, fitted parameters, or self-citations are used to derive results; the central output is the curated repository itself. The abstract and description contain no load-bearing steps that reduce to self-definition or fitted inputs. This matches the default non-circular case for compilation papers.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption A single DFT functional and convergence settings produce comparable enthalpies across chemically diverse structures
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
all retained structures underwent re-optimization under a unified density functional theory (DFT) framework, with continuous enthalpy curves systematically generated specifically for the elemental phases
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
HPCSD establishes a robust infrastructure to accelerate experimental phase identification, facilitate cross-study thermodynamic comparisons
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
L. Zhang, Y . Wang, J. Lv, Y . Ma, Materials discovery at high pressures, Nature Reviews Materials 2 (2017) 17005. doi:10.1038/natrevmats.2017.5
-
[2]
H. Wang, J. S. Tse, K. Tanaka, T. Iitaka, Y . Ma, Su- perconductive sodalite-like clathrate calcium hydride at high pressures, Proceedings of the National Academy of Sciences 109 (2012) 6463–6466. doi:10.1073/pnas. 1118168109
-
[3]
H. Liu, I. I. Naumov, R. Hoffmann, N. W. Ashcroft, R. J. Hemley, Potential high-Tc superconducting lanthanum and yttrium hydrides at high pressure, Proceedings of the National Academy of Sciences 114 (2017) 6990–6995. doi:10.1073/pnas.1704505114
-
[4]
F. Peng, Y . Sun, C. J. Pickard, R. J. Needs, Q. Wu, Y . Ma, Hydrogen Clathrate Structures in Rare Earth Hydrides at High Pressures: Possible Route to Room-Temperature Superconductivity, Physical Review Letters 119 (2017) 107001. doi:10.1103/physrevlett.119.107001
-
[5]
X.-L. He, W. Zhao, Y . Xie, A. Hermann, R. J. Hem- ley, H. Liu, Y . Ma, Predicted hot superconductivity in LaSc2H24 under pressure, Proceedings of the Na- tional Academy of Sciences 121 (2024) e2401840121. doi:10.1073/pnas.2401840121
-
[6]
L. Ma, K. Wang, Y . Xie, X. Yang, Y . Wang, M. Zhou, H. Liu, X. Yu, Y . Zhao, H. Wang, G. Liu, Y . Ma, High-Temperature Superconducting Phase in Clathrate Calcium Hydride CaH6 up to 215 K at a Pressure of 172 GPa, Physical Review Letters 128 (2022) 167001. doi:10.1103/physrevlett.128.167001. 5
-
[7]
Z. Li, X. He, C. Zhang, X. Wang, S. Zhang, Y . Jia, S. Feng, K. Lu, J. Zhao, J. Zhang, B. Min, Y . Long, R. Yu, L. Wang, M. Ye, Z. Zhang, V . Prakapenka, S. Chariton, P. A. Ginsberg, J. Bass, S. Yuan, H. Liu, C. Jin, Supercon- ductivity above 200 K discovered in superhydrides of cal- cium, Nature Communications 13 (2022) 2863. doi:10. 1038/s41467-022-3045...
-
[8]
A. P. Drozdov, P. P. Kong, V . S. Minkov, S. P. Besedin, M. A. Kuzovnikov, S. Mozaffari, L. Balicas, F. F. Bal- akirev, D. E. Graf, V . B. Prakapenka, E. Greenberg, D. A. Knyazev, M. Tkacz, M. I. Eremets, Supercon- ductivity at 250 K in lanthanum hydride under high pressures, Nature 569 (2019) 528–531. doi:10.1038/ s41586-019-1201-8
work page 2019
- [10]
-
[11]
A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder, K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials in- novation, APL Materials 1 (2013) 011002. doi:10.1063/ 1.4812323
work page 2013
-
[12]
J. E. Saal, S. Kirklin, M. Aykol, B. Meredig, C. Wolver- ton, Materials Design and Discovery with High- Throughput Density Functional Theory: The Open Quan- tum Materials Database (OQMD), JOM 65 (2013) 1501–
work page 2013
-
[13]
doi:10.1007/s11837-013-0755-4
-
[14]
F. Giannessi, S. D. Cataldo, S. Saha, L. Boeri, A database of high-pressure crystal structures from hydrogen to lan- thanum, Scientific Data 11 (2024) 766. doi:10.1038/ s41597-024-03447-1
work page 2024
-
[15]
X. Wang, Y . Wang, M. Miao, X. Zhong, J. Lv, T. Cui, J. Li, L. Chen, C. J. Pickard, Y . Ma, Cagelike Dia- mondoid Nitrogen at High Pressures, Physical Review Letters 109 (2012) 175502. doi:10.1103/physrevlett. 109.175502
-
[16]
M. Miao, Y . Sun, E. Zurek, H. Lin, Chemistry under high pressure, Nature Reviews Chemistry 4 (2020) 508–527. doi:10.1038/s41570-020-0213-0
-
[17]
Y . Ma, M. Eremets, A. R. Oganov, Y . Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle, V . Prakapenka, Transparent dense sodium, Nature 458 (2009) 182–185. doi:10.1038/nature07786
-
[18]
C. V . Storm, S. E. Finnegan, J. D. McHardy, M. J. Duff, M. I. McMahon, S. G. MacLeod, E. Plekhanov, C. Weber, Unified High-Pressure Phase-Transition Se- quence in the f-Electron Metals: oF16→oF8 Transition in Terbium, Physical Review Letters 135 (2025) 136101. doi:10.1103/r3zx-k97x
-
[19]
J. C. Duthie, D. G. Pettifor, Correlation between d-Band Occupancy and Crystal Structure in the Rare Earths, Phys- ical Review Letters 38 (1977) 564–567. doi:10.1103/ physrevlett.38.564
work page 1977
-
[20]
B. Johansson, Structural and electronic relationships between the lanthanide and actinide elements, Hy- perfine Interactions 128 (2000) 41–66. doi:10.1023/a: 1012667128586
work page doi:10.1023/a: 2000
-
[21]
U. Häussermann, High-Pressure Structural Trends of Group 15 Elements: Simple Packed Structures versus Complex Host–Guest Arrangements, Chemistry – A Eu- ropean Journal 9 (2003) 1471–1478. doi:10.1002/chem. 200390166. 6
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.