Chemical Interpretation of Time-Dependent Coupled-Cluster Theory
Pith reviewed 2026-05-19 22:46 UTC · model grok-4.3
The pith
Time-dependent coupled-cluster theory gains chemical interpretation through expansion of its wavefunctions in a Slater-determinant basis to produce time-dependent configuration weights.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
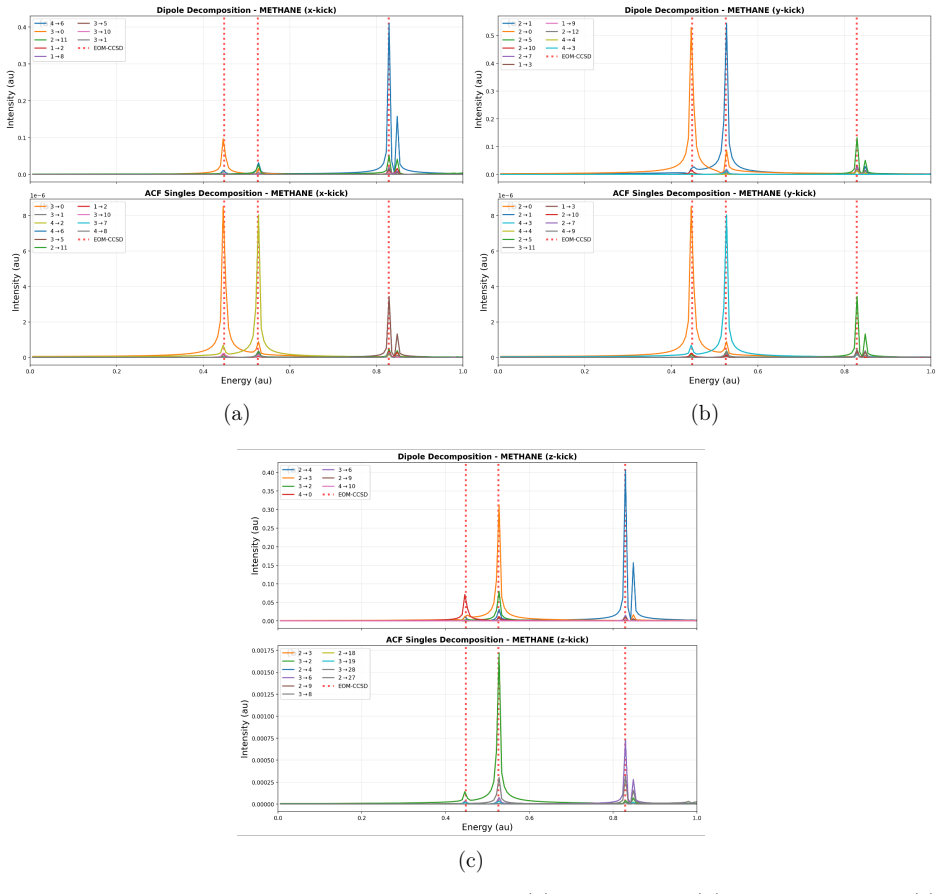
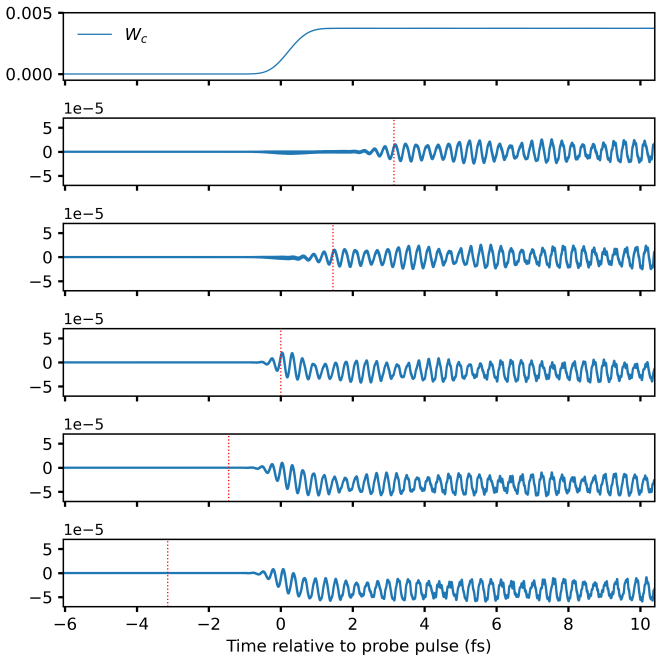
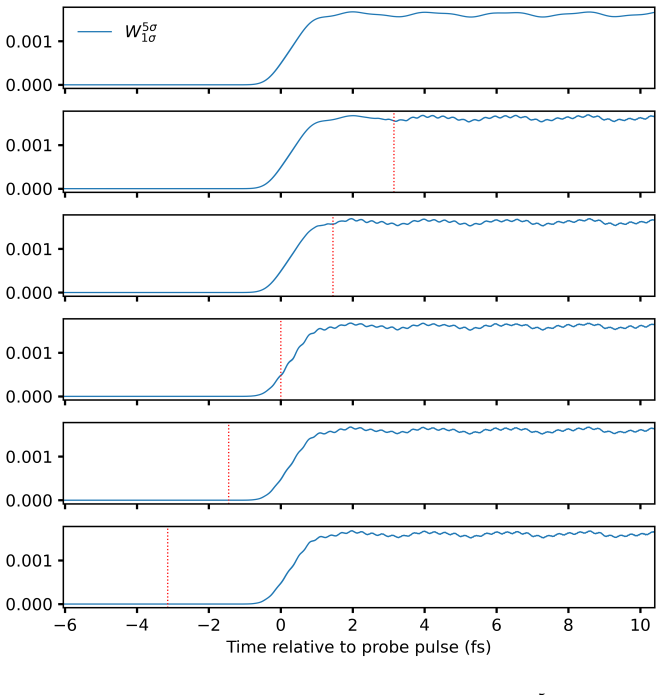
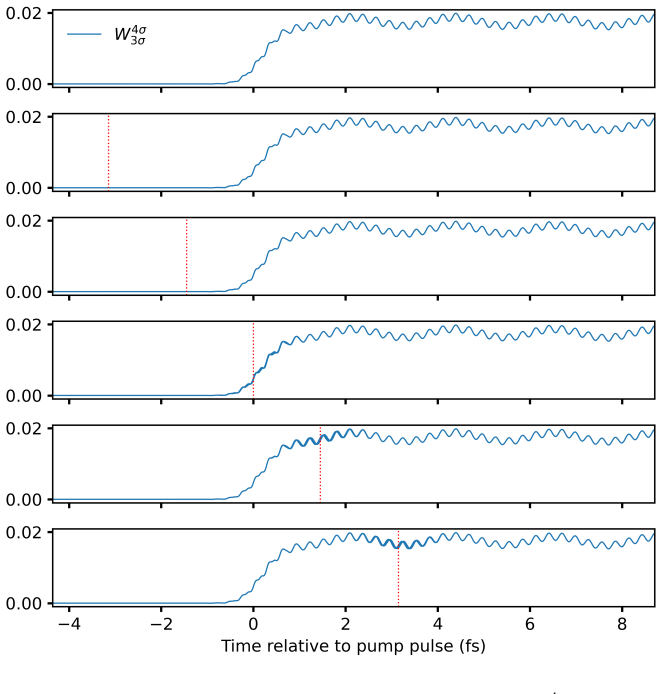
At the time-dependent coupled-cluster singles-and-doubles level the left and right wavefunctions are expanded in the full Slater-determinant basis to obtain time-dependent configuration weights. These weights decompose the electric-dipole moment and autocorrelation function, allowing each feature in a linear absorption spectrum to be assigned to a dominant orbital transition. The resulting assignments for valence lines in HF, H2O, NH3, and CH4 match those obtained from equation-of-motion coupled-cluster singles-and-doubles theory, and the same procedure is applied to core-level excitations and to time-resolved processes such as impulsive x-ray Raman scattering in neon and pump-probe spectra.
What carries the argument
Time-dependent configuration weights obtained by expanding the left and right coupled-cluster functions in the Slater-determinant basis, which quantify the instantaneous contribution of each determinant and enable decomposition of dipole and autocorrelation signals.
If this is right
- Valence absorption lines in HF, H2O, NH3, and CH4 receive explicit orbital-transition labels that agree with equation-of-motion coupled-cluster results.
- Core-level excitations in HF, H2O, and NH3 can be assigned by the same decomposition of the dipole moment.
- Population dynamics during impulsive stimulated x-ray Raman scattering in the neon atom become directly visible through the time evolution of the weights.
- Transient pump-probe spectra of HF can be interpreted by tracking how individual determinants contribute at each delay time.
Where Pith is reading between the lines
- The same determinant expansion could be applied to time-dependent simulations that include higher-order excitations to check whether the assignments remain stable.
- Direct comparison of these weights with frequency-domain response functions might reveal how time-domain truncation errors translate into spectral features.
- The method offers a route to interpret dynamics in systems where static excited-state methods become computationally expensive.
Load-bearing premise
The configuration weights derived from the determinant expansion correspond to the actual dominant orbital-excitation character of the dynamics rather than being an artifact of the chosen reference determinant or the truncation level.
What would settle it
If the orbital-transition assignments produced by the time-dependent configuration weights differ systematically from the assignments obtained by equation-of-motion coupled-cluster singles-and-doubles theory on the same four ten-electron molecules, the chemical interpretation would be shown to be unreliable.
Figures












read the original abstract
While providing a highly accurate framework for simulating laser-induced many-electron dynamics in atom and molecules, including linear and nonlinear steady-state and transient absorption spectra, time-dependent coupled-cluster theory does not offer a straightforward interpretation in chemical terms. This should be contrasted with conventional time-independent equation-of-motion coupled-cluster or frequency-dependent response models where a simple eigenvector analysis readily reveals the dominant orbital-excitation character of individual excited states. We fill this gap by expanding the left and right coupled-cluster functions in Slater-determinant basis, thus allowing for a time-dependent generalization of configuration weights that can be used to track populations throughout a simulation. The same expansions are used to decompose the time-dependent electric-dipole moment and autocorrelation function, providing a computationally straightforward approach to the assignment of absorption peaks to orbital transitions for single-reference systems. At the time-dependent coupled-cluster singles-and-doubles level of theory, we demonstrate the power of the proposed methodology by assigning valence lines in the linear absorption spectra of four ten-electron molecules (HF, H2O, NH3, and CH4) with different point-group symmetries, validating the assignment by comparison with equation-of-motion coupled-cluster singles-and-doubles theory. In addition, core-level excitations are assigned for HF, H2O, and NH3. Finally, the usefulness of time-dependent configuration weights is illustrated by applications to an impulsive stimulated x-ray Raman scattering process in the Ne atom and to a transient pump-probe spectrum of the HF molecule.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a chemical interpretation for time-dependent coupled-cluster (TD-CC) theory by expanding the left and right CC vectors in the Slater-determinant basis. This yields time-dependent configuration weights that are used to decompose the time-dependent dipole moment and autocorrelation function, enabling assignment of absorption peaks to specific orbital transitions. The method is demonstrated at the CCSD level for valence-line assignments in HF, H2O, NH3, and CH4 (validated against EOM-CCSD), core excitations in three of those molecules, and applications to impulsive stimulated x-ray Raman scattering in Ne and a transient pump-probe spectrum of HF.
Significance. If the configuration weights prove robust against truncation artifacts, the approach would supply a practical, post-processing route to orbital-level insight in TD-CC simulations of laser-driven dynamics. This would complement the high accuracy of TD-CC for spectra and transients while addressing the interpretability gap relative to EOM-CC or response theory, with direct relevance to attosecond and nonlinear spectroscopies.
major comments (2)
- [§4.1] §4.1 (valence assignments): The validation consists of a qualitative statement that the TD-CCSD assignments match EOM-CCSD; no quantitative metrics (e.g., overlap of dominant configurations, percentage agreement across the four molecules, or energy deviations) are supplied. Because the central demonstration rests on this comparison, the absence of such numbers leaves the strength of the agreement unassessed.
- [§4.2] §4.2 (core excitations): Both the TD-CCSD weights and the EOM-CCSD reference employ the identical HF reference and CCSD truncation. For core states, where single-reference and truncation errors are known to be larger, this shared bias could produce matching but spurious orbital assignments; an explicit test against a higher-level method or experimental peak positions is needed to substantiate the claim.
minor comments (2)
- [Theory section] The definition of the time-dependent weights (Eq. (X) in the theory section) should explicitly state the normalization convention used for the left and right vectors to avoid ambiguity when comparing populations across time steps.
- [Figure captions] Figure captions for the decomposed spectra should indicate the threshold used to label a configuration as 'dominant' so that readers can reproduce the assignments.
Simulated Author's Rebuttal
We thank the referee for their supportive summary and recommendation of minor revision. We respond point by point to the major comments below, indicating where revisions will be made to strengthen the manuscript.
read point-by-point responses
-
Referee: [§4.1] §4.1 (valence assignments): The validation consists of a qualitative statement that the TD-CCSD assignments match EOM-CCSD; no quantitative metrics (e.g., overlap of dominant configurations, percentage agreement across the four molecules, or energy deviations) are supplied. Because the central demonstration rests on this comparison, the absence of such numbers leaves the strength of the agreement unassessed.
Authors: We agree that quantitative metrics would provide a clearer assessment of the agreement. In the revised manuscript we will add a table (or supplementary material) reporting, for each valence peak in HF, H2O, NH3, and CH4, the dominant configurations and their weights from both TD-CCSD and EOM-CCSD, the percentage of matching dominant assignments, and the mean absolute deviation in excitation energies. revision: yes
-
Referee: [§4.2] §4.2 (core excitations): Both the TD-CCSD weights and the EOM-CCSD reference employ the identical HF reference and CCSD truncation. For core states, where single-reference and truncation errors are known to be larger, this shared bias could produce matching but spurious orbital assignments; an explicit test against a higher-level method or experimental peak positions is needed to substantiate the claim.
Authors: We acknowledge the risk of shared bias for core excitations. A systematic comparison against CCSDT or higher is computationally prohibitive within the present scope and would require a separate study. However, we will revise the manuscript to include direct comparisons of the assigned TD-CCSD core excitation energies with available experimental values for HF, H2O, and NH3, and we will add an explicit discussion of the limitations of the CCSD truncation for core states. revision: partial
Circularity Check
No circularity: time-dependent configuration weights introduced as independent extension with external EOM-CCSD validation
full rationale
The paper introduces an expansion of left and right TD-CC functions in the Slater-determinant basis to generate time-dependent configuration weights, then uses these to decompose the dipole moment and autocorrelation function for spectral assignment. This step is presented as a new methodological proposal rather than a derivation that reduces to prior equations or fitted parameters within the same calculation. Validation proceeds via direct comparison to separate EOM-CCSD calculations on HF, H2O, NH3, and CH4, which operate as an independent benchmark at the same truncation level but do not constitute a self-referential fit or redefinition. No self-definitional loops, fitted inputs relabeled as predictions, or load-bearing self-citations appear in the chain; the central claim rests on standard CC machinery plus the proposed expansion, which remains falsifiable against external references.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Time-dependent coupled-cluster singles-and-doubles provides a sufficiently accurate description of the valence and core excitations studied.
- ad hoc to paper The Slater-determinant expansion of the left and right CC vectors produces weights whose time evolution tracks orbital populations without significant truncation artifacts.
Reference graph
Works this paper leans on
-
[1]
Suzuki, H. Electronic absorption spectra and geometry of organic molecules: An application of molecular orbital theory ; Academic Press: New York, 1967
work page 1967
-
[2]
Molecular Electronic-Structure Theory ; Wiley: Chichester, 2013
Helgaker, T.; J rgensen, P.; Olsen, J. Molecular Electronic-Structure Theory ; Wiley: Chichester, 2013
work page 2013
-
[3]
English, N. J.; Waldron, C. J. Perspectives on external electric fields in molecular simulation: progress, prospects and challenges . Phys. Chem. Chem. Phys. 2015, 17, 12407--12440
work page 2015
-
[4]
Goings, J. J.; Lestrange, P. J.; Li, X. Real-time time-dependent electronic structure theory . WIREs Comput. Mol. Sci. 2018, 8, e1341
work page 2018
-
[5]
The quantum chemistry of attosecond molecular science
Palacios, A.; Martín, F. The quantum chemistry of attosecond molecular science. WIREs Comput. Mol. Sci. 2020, 10, e1430
work page 2020
-
[6]
Li, X.; Govind, N.; Isborn, C.; DePrince III, A. E.; Lopata, K. Real-Time Time-Dependent Electronic Structure Theory . Chem. Rev. 2020, 120, 9951--9993
work page 2020
-
[7]
Real-time TD-DFT with classical ion dynamics: Methodology and applications
Kolesov, G.; Grånäs, O.; Hoyt, R.; Vinichenko, D.; Kaxiras, E. Real-time TD-DFT with classical ion dynamics: Methodology and applications . J. Chem. Theory Comput. 2016, 12, 466--476
work page 2016
-
[8]
Hoodbhoy, P.; Negele, J. W. Time-dependent coupled-cluster approximation to nuclear dynamics. II. General formulation . Phys. Rev. C 1979, 19, 1971--1982
work page 1979
-
[9]
Variational principles and linked-cluster exp S expansions for static and dynamic many-body problems
Arponen, J. Variational principles and linked-cluster exp S expansions for static and dynamic many-body problems . Ann. Phys. 1983, 151, 311--382
work page 1983
-
[10]
Pedersen, T. B.; Koch, H. On the time-dependent Lagrangian approach in quantum chemistry . J. Chem. Phys. 1998, 108, 5194--5204
work page 1998
-
[11]
Huber, C.; Klamroth, T. Explicitly time-dependent coupled cluster singles doubles calculations of laser-driven many-electron dynamics . J. Chem. Phys. 2011, 134, 054113
work page 2011
-
[12]
Ab initio quantum dynamics using coupled-cluster
Kvaal, S. Ab initio quantum dynamics using coupled-cluster . J. Chem. Phys. 2012, 136, 194109
work page 2012
-
[13]
Nascimento, D. R.; DePrince III, A. E. Linear absorption spectra from explicitly time-dependent equation-of-motion coupled-cluster theory . J. Chem. Theory Comput. 2016, 12, 5834--5840
work page 2016
-
[14]
Sato, T.; Pathak, H.; Orimo, Y.; Ishikawa, K. L. Time-dependent optimized coupled-cluster method for multielectron dynamics . J. Chem. Phys. 2018, 148, 051101
work page 2018
-
[15]
Pedersen, T. B.; Kvaal, S. Symplectic integration and physical interpretation of time-dependent coupled-cluster theory . J. Chem. Phys. 2019, 150, 144106
work page 2019
-
[16]
Hansen, M. B.; Madsen, N. K.; Zoccante, A.; Christiansen, O. Time-dependent vibrational coupled cluster theory: Theory and implementation at the two-mode coupling level . J. Chem. Phys. 2019, 151, 154116
work page 2019
-
[17]
Koulias, L. N.; Williams-Young, D. B.; Nascimento, D. R.; Deprince, A. E.; Li, X. Relativistic Real-Time Time-Dependent Equation-of-Motion Coupled-Cluster . J. Chem. Theory Comput. 2019, 15, 6617--6624
work page 2019
-
[18]
Skeidsvoll, A. S.; Balbi, A.; Koch, H. Time-dependent coupled-cluster theory for ultrafast transient-absorption spectroscopy . Phys. Rev. A 2020, 102, 023115
work page 2020
-
[19]
Wang, Z.; Peyton, B. G.; Crawford, T. D. Accelerating real-time coupled cluster methods with single-precision arithmetic and adaptive numerical integration . J. Chem. Theory Comput. 2022, 18, 5479--5491
work page 2022
-
[20]
Ofstad, B. S.; Aurbakken, E.; Sch yen, . S.; Kristiansen, H. E.; Kvaal, S.; Pedersen, T. B. Time-dependent coupled-cluster theory . WIREs Comput. Mol. Sci. 2023, 13, e1666
work page 2023
-
[21]
Kvaal, S.; Fredheim, H. R.; Højlund, M. G.; Pedersen, T. B. Time-Dependent Bivariational Principle: Theoretical Foundation for Real-Time Propagation Methods of Coupled-Cluster Type . J. Phys. Chem. A 2025, 129, 3508--3521
work page 2025
-
[22]
Wang, Z.; Kristiansen, H. E.; Pedersen, T. B.; Crawford, T. D. Real-Time Coupled Cluster Theory with Approximate Triples . J. Phys. Chem. A 2025, 129, 1908--1927
work page 2025
-
[23]
Crawford, T. D.; Kumar, A.; Bazanté, A. P.; Di Remigio, R. Reduced-scaling coupled cluster response theory: Challenges and opportunities . WIREs Comput. Mol. Sci. 2019, 9, e1406
work page 2019
-
[24]
Peyton, B. G.; Wang, Z.; Crawford, T. D. Reduced Scaling Real-Time Coupled Cluster Theory . J. Phys. Chem. A 2023, 127, 8486--8499
work page 2023
-
[25]
Hauge, E. S. Extrapolating the Electric Dipole Moment . M.Sc.\ thesis, 2021; University of Oslo, https://hdl.handle.net/11250/4170547
work page 2021
-
[26]
E.; Konecny, L.; Kadek, M.; Repisky, M.; Pedersen, T
Hauge, E.; Kristiansen, H. E.; Konecny, L.; Kadek, M.; Repisky, M.; Pedersen, T. B. Cost-Efficient High-Resolution Linear Absorption Spectra through Extrapolating the Dipole Moment from Real-Time Time-Dependent Electronic-Structure Theory . J. Chem. Theory Comput. 2023, 19, 7764--7775
work page 2023
-
[27]
Super-resolution techniques to simulate electronic spectra of large molecular systems
Kick, M.; Alexander, E.; Beiersdorfer, A.; Van Voorhis, T. Super-resolution techniques to simulate electronic spectra of large molecular systems . Nat. Commun. 2024, 15, 8001
work page 2024
-
[28]
Stanton, J. F.; Bartlett, R. J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties . J. Chem. Phys. 1993, 98, 7029--7039
work page 1993
-
[29]
Pedersen, T. B.; Kristiansen, H. E.; Bodenstein, T.; Kvaal, S.; Sch yen, . S. Interpretation of Coupled-Cluster Many-Electron Dynamics in Terms of Stationary States . J. Chem. Theory Comput. 2021, 17, 388--404
work page 2021
-
[30]
E.; Kvernmoen, H.; Kvaal, S.; Pedersen, T
Kristiansen, H. E.; Kvernmoen, H.; Kvaal, S.; Pedersen, T. B. Configuration Weights in Coupled-Cluster Theory . J. Phys. Chem. A 2025, 129, 2638--2654
work page 2025
-
[31]
Wibowo, M.; Irons, T. J.; Teale, A. M. Modeling ultrafast electron dynamics in strong magnetic fields using real-time time-dependent electronic structure methods. J. Chem. Theory Comput. 2021, 17, 2137--2165
work page 2021
-
[32]
Repisky, M.; Konecny, L.; Kadek, M.; Komorovsky, S.; Malkin, O. L.; Malkin, V. G.; Ruud, K. Excitation energies from real-time propagation of the four-component Dirac--Kohn--Sham equation . J. Chem. Theory Comput. 2015, 11, 980--991
work page 2015
-
[33]
Accelerated broadband spectra using transition dipole decomposition and Pad \'e approximants
Bruner, A.; LaMaster, D.; Lopata, K. Accelerated broadband spectra using transition dipole decomposition and Pad \'e approximants . J. Chem. Theory Comput. 2016, 12, 3741--3750
work page 2016
-
[34]
Abraham, V.; Senapati, P.; Pathak, H.; Peng, B. Elucidating many-body effects in molecular core spectra through real-time approaches: Efficient classical approximations and a quantum perspective. J. Chem. Phys. 2026, 164
work page 2026
-
[35]
Time-Dependent Schr \"o dinger Equation
Scrinzi, A. Time-Dependent Schr \"o dinger Equation . Attosecond and XUV Physics: Ultrafast Dynamics and Spectroscopy 2014, 257--292
work page 2014
-
[36]
Aurbakken, E.; Ofstad, B. S.; Kristiansen, H. E.; Schøyen, . S.; Kvaal, S.; Sørensen, L. K.; Lindh, R.; Pedersen, T. B. Transient spectroscopy from time-dependent electronic-structure theory without multipole expansions. Phys. Rev. A 2024, 109, 013109
work page 2024
-
[37]
Bracewell, R. N. The Fourier Transform . Scientific American 1989, 260, 86--95
work page 1989
-
[38]
Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen . J. Chem. Phys. 1989, 90, 1007--1023
work page 1989
-
[39]
Crawford, T. D.; Peyton, B. G.; Wang, Z.; Madriaga, J. PyCC---A Python-based coupled cluster implementation. 2026; https://github.com/CrawfordGroup/pycc, (access date: 2026-01-28)
work page 2026
-
[40]
Smith, D. G.; Burns, L. A.; Simmonett, A. C.; Parrish, R. M.; Schieber, M. C.; Galvelis, R.; Kraus, P.; Kruse, H.; Di Remigio, R.; Alenaizan, A. et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry . J. Chem. Phys. 2020, 152
work page 2020
-
[41]
Coriani, S.; Koch, H. X-ray absorption spectra and core-ionization potentials within a core-valence separated coupled cluster framework . J. Chem. Phys. 2015, 143, 181103
work page 2015
-
[42]
Balbi, A.; Skeidsvoll, A. S.; Koch, H. Coupled Cluster Simulation of Impulsive Stimulated X-ray Raman Scattering . J. Phys. Chem. A 2023, 127, 8676--8684
work page 2023
-
[43]
Trigonometric pulse envelopes for laser-induced quantum dynamics
Barth, I.; Lasser, C. Trigonometric pulse envelopes for laser-induced quantum dynamics. J. Phys. B 2009, 42, 235101
work page 2009
-
[44]
Balbi, A.; Koch, H. Private communication. There is a misprint in Ref. balbi_coupled_2023 . The correct value used for in that work is not 0.5\, a.u. but 5\, a.u
-
[45]
Kendall, R. A.; Dunning, T. H.; Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions . J. Chem. Phys. 1992, 96, 6796--7006
work page 1992
-
[46]
Woon, D. E.; Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. V. Core‐valence basis sets for boron through neon . J. Chem. Phys. 1995, 103, 4572--4585
work page 1995
-
[47]
Aurbakken, E.; Fredly, K. H.; Kristiansen, H. E.; Kvaal, S.; Myhre, R. H.; Ofstad, B. S.; Pedersen, T. B.; Sch yen, . S.; Sutterud, H.; Winther-Larsen, S. G. HyQD: Hylleraas Quantum Dynamics . 2026; https://github.com/HyQD, (access date: 2026-01-28)
work page 2026
-
[48]
Sun, Q.; Berkelbach, T. C.; Blunt, N. S.; Booth, G. H.; Guo, S.; Li, Z.; Liu, J.; McClain, J. D.; Sayfutyarova, E. R.; Sharma, S. et al. PySCF: the Python‐based simulations of chemistry framework . WIREs Comput. Mol. Sci. 2018, 8, e1340
work page 2018
-
[49]
Sun, Q.; Zhang, X.; Banerjee, S.; Bao, P.; Barbry, M.; Blunt, N. S.; Bogdanov, N. A.; Booth, G. H.; Chen, J.; Cui, Z.-H. et al. Recent developments in the PySCF program package . J. Chem. Phys. 2020, 153, 024109
work page 2020
-
[50]
Kvaal, S.; Lasser, C.; Pedersen, T. B.; Adamowicz, L. No need for a grid: Adaptive fully-flexible gaussians for the time-dependent Schrödinger equation . 2023; arXiv:2207.00271 [quant-ph]
-
[51]
Schrader, S. E.; Kristiansen, H. E.; Pedersen, T. B.; Kvaal, S. Time evolution as an optimization problem: The hydrogen atom in strong laser fields in a basis of time-dependent Gaussian wave packets . J. Chem. Phys. 2024, 161, 044105
work page 2024
-
[52]
P.; Adamowicz, L.; Pedersen, T
Woźniak, A. P.; Adamowicz, L.; Pedersen, T. B.; Kvaal, S. Gaussians for Electronic and Rovibrational Quantum Dynamics . J. Phys. Chem. A 2024, 128, 3659--3671
work page 2024
-
[53]
Schrader, S. E.; Pedersen, T. B.; Kvaal, S. Multidimensional quantum dynamics with explicitly correlated Gaussian wave packets using Rothe’s method . J. Chem. Phys. 2025, 162, 024109
work page 2025
-
[54]
P.; Adamowicz, L.; Pedersen, T
Woźniak, A. P.; Adamowicz, L.; Pedersen, T. B.; Kvaal, S. Rothe Time Propagation for Coupled Electronic and Rovibrational Quantum Dynamics . J. Phys. Chem. A 2025, 129, 5391--5404
work page 2025
-
[55]
Schrader, S. E.; Kristiansen, H. E.; Pedersen, T. B.; Kvaal, S. Time-Dependent Gaussian Basis Sets for Many-Body Systems Using Rothe’s Method: A Mean-Field Study . J. Chem. Theory Comput. 2025, 21, 8490--8508
work page 2025
-
[56]
Wu, M.; Chen, S.; Camp, S.; Schafer, K. J.; Gaarde, M. B. Theory of strong-field attosecond transient absorption. J. Phys. B 2016, 49, 062003 mcitethebibliography document
work page 2016
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.