SPARC-atomSFE: Spectral finite-element package for atomic structure calculations in density functional theory
Pith reviewed 2026-05-20 02:13 UTC · model grok-4.3
The pith
The SPARC-atomSFE package performs atomic Kohn-Sham DFT calculations to 1 micro-Hartree accuracy across local to nonlocal functionals using spectral finite elements.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
SPARC-atomSFE solves the atomic Kohn-Sham equations by means of a spectral finite-element discretization on an adaptive radial grid. Element nodes follow the Legendre-Gauss-Lobatto distribution, the basis consists of high-order C0-continuous Lagrange polynomials, and integrals are evaluated by Gauss-Legendre quadrature. The code supports both all-electron and pseudopotential calculations and implements local, semilocal, hybrid, and random-phase-approximation functionals, the latter via either the generalized Kohn-Sham or optimized-effective-potential route. Systematic convergence studies identify the parameters needed for target accuracy, and representative calculations confirm agreement to
What carries the argument
Adaptive spectral finite-element discretization of the radial Kohn-Sham problem using high-order Lagrange polynomials on Legendre-Gauss-Lobatto nodes together with Gauss-Legendre quadrature.
If this is right
- Both generalized Kohn-Sham and optimized-effective-potential routes become available for eigenvalue-dependent functionals such as hybrids and RPA.
- Users can reach any target accuracy by systematically refining the radial grid or raising the polynomial degree.
- All-electron and norm-conserving pseudopotential results are obtained inside the same code and discretization framework.
- Atomic energies serve as clean benchmarks for developing or validating new exchange-correlation approximations.
Where Pith is reading between the lines
- The adaptive radial mesh could be generalized to multi-center molecular geometries by allowing element refinement around each nucleus.
- Direct timing comparisons against established atomic codes that use finite differences or B-splines would reveal any efficiency gains from the spectral-element approach.
- The OEP implementation for nonlocal functionals opens a route to other advanced many-body corrections that depend on orbital eigenvalues.
Load-bearing premise
The chosen adaptive grid, polynomial degree, and quadrature rules remain free of significant numerical artifacts for the tested atoms and functionals once the reported convergence parameters are used.
What would settle it
A total-energy calculation for the neon atom in the LDA functional that deviates from the accepted literature value by more than 1 micro-Hartree when the package is run at the convergence settings stated in the paper.
Figures




read the original abstract
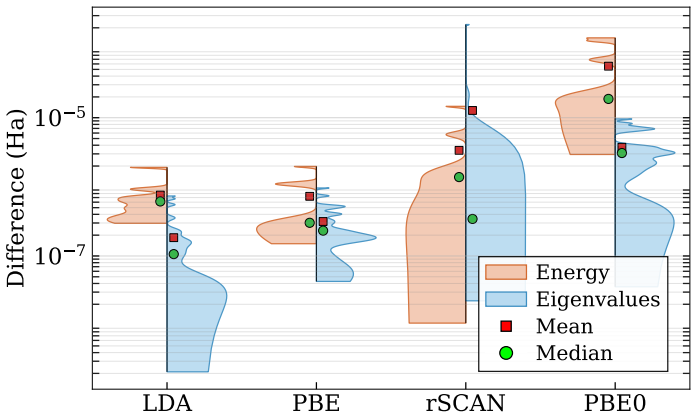
We present SPARC-atomSFE, a spectral finite-element package for accurate and efficient atomic structure calculations within the framework of Kohn-Sham density functional theory. The package supports both all-electron and norm conserving pseudopotential calculations across a comprehensive hierarchy of exchange-correlation approximations, spanning local, semilocal, and nonlocal functionals. The latter includes hybrid functionals and the many-body random phase approximation, for which we implement both the generalized Kohn-Sham approach and the optimized effective potential (OEP) method, with OEP necessary for eigenvalue-dependent functionals. Spatial discretization is based on an adaptive grid with element nodes distributed according to the Legendre--Gauss--Lobatto scheme, high-order $C^{0}$-continuous Lagrange polynomial basis functions, and Gauss--Legendre quadrature for numerical integration. We present systematic convergence studies and identify the computational parameters required to achieve target accuracies. We validate the accuracy of SPARC-atomSFE through representative calculations spanning the various exchange-correlations approximations, obtaining results that generally agree with values in the literature to within $1~\mu\text{Ha}$ or better.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents SPARC-atomSFE, a spectral finite-element code for Kohn-Sham DFT atomic calculations. It supports all-electron and norm-conserving pseudopotential treatments across local, semilocal, hybrid, and RPA functionals, implementing both generalized Kohn-Sham and optimized effective potential (OEP) approaches for eigenvalue-dependent cases. Discretization relies on an adaptive radial grid with Legendre-Gauss-Lobatto nodes, C0 Lagrange polynomials of high order, and Gauss-Legendre quadrature. Systematic convergence studies are used to select parameters for target accuracy, and the code is validated by direct comparison to literature total energies and eigenvalues, with reported agreement generally within 1 μHa or better.
Significance. If the reported numerical accuracies are confirmed, the package supplies a useful, reproducible platform for high-precision atomic reference data, especially for advanced nonlocal functionals that require OEP. The combination of adaptive high-order elements with broad functional coverage and explicit convergence diagnostics adds value for method benchmarking and functional testing in atomic DFT.
major comments (1)
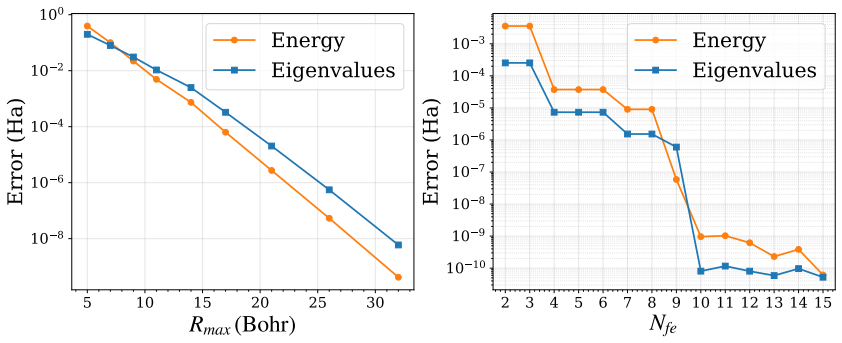
- [Convergence studies and validation sections] The central accuracy claim (agreement to 1 μHa) rests on the assertion that the chosen adaptive-grid, polynomial degree, and quadrature settings are fully converged. The manuscript should include explicit refinement tables or figures (e.g., energy change upon increasing polynomial degree by 2 or halving the adaptive mesh spacing) for the most demanding cases: all-electron runs near the nuclear cusp and OEP calculations with eigenvalue-dependent functionals. Without such data, it is difficult to confirm that discretization error lies well below the target threshold for every functional class shown.
minor comments (4)
- [Abstract] Abstract, line 3: 'exchange-correlations approximations' should read 'exchange-correlation approximations'.
- [Discretization section] The notation for the radial adaptive grid and the mapping from reference element to physical element should be stated once in a single equation block rather than repeated in prose.
- [Figures] Figure captions for the convergence plots would benefit from explicit mention of the atom, functional, and quantity (total energy or eigenvalue) being plotted, together with the final parameter set used for the validation table.
- [Results] A short paragraph comparing wall-time or iteration counts against at least one existing atomic DFT code (e.g., a finite-difference or Gaussian-basis implementation) would help readers assess the practical efficiency of the spectral-element approach.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of the manuscript and the constructive recommendation for minor revision. We address the major comment below.
read point-by-point responses
-
Referee: [Convergence studies and validation sections] The central accuracy claim (agreement to 1 μHa) rests on the assertion that the chosen adaptive-grid, polynomial degree, and quadrature settings are fully converged. The manuscript should include explicit refinement tables or figures (e.g., energy change upon increasing polynomial degree by 2 or halving the adaptive mesh spacing) for the most demanding cases: all-electron runs near the nuclear cusp and OEP calculations with eigenvalue-dependent functionals. Without such data, it is difficult to confirm that discretization error lies well below the target threshold for every functional class shown.
Authors: We agree that additional explicit refinement data would further strengthen the convergence evidence presented in the manuscript. While systematic convergence studies were performed to identify parameters achieving the target accuracy, we will incorporate new tables and figures in the revised manuscript. These will report energy changes upon increasing the polynomial degree by 2 and upon halving the adaptive mesh spacing, specifically for the most demanding cases of all-electron calculations near the nuclear cusp and OEP calculations involving eigenvalue-dependent functionals. This will explicitly demonstrate that discretization errors remain well below the 1 μHa threshold across the functional classes considered. revision: yes
Circularity Check
No circularity: SPARC-atomSFE is a software implementation validated against independent external literature
full rationale
The manuscript presents a spectral finite-element code for atomic Kohn-Sham DFT calculations. It adopts standard discretization (adaptive radial grid, Legendre-Gauss-Lobatto nodes, C0 Lagrange polynomials, Gauss-Legendre quadrature) and reports systematic convergence studies followed by direct numerical comparison of total energies and eigenvalues to previously published reference values across local, semilocal, hybrid, and RPA functionals. No result is obtained by fitting a parameter to a subset of the same data and then relabeling it a prediction; no central premise rests on a self-citation chain whose own justification is internal to the present work; and the validation step is explicitly benchmarked against external literature rather than closed under the code's own outputs. The derivation chain is therefore self-contained and non-circular.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Kohn-Sham density functional theory provides a sufficiently accurate framework for the atomic systems and properties considered.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Spatial discretization is based on an adaptive grid with element nodes distributed according to the Legendre–Gauss–Lobatto scheme, high-order C0-continuous Lagrange polynomial basis functions, and Gauss–Legendre quadrature for numerical integration.
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We validate the accuracy of SPARC-atomSFE through representative calculations spanning the various exchange-correlations approximations, obtaining results that generally agree with values in the literature to within 1 μHa or better.
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
W. Kohn, L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev. 140 (4A) (1965) A1133
work page 1965
-
[2]
P. Hohenberg, W. Kohn, Inhomogeneous electron gas, Phys. Rev. 136 (3B) (1964) 864
work page 1964
-
[3]
J. P. Perdew, K. Schmidt, Jacob’s ladder of density functional approximations for the exchange- correlation energy, AIP Conf. Proc. 577 (1) (2001) 1–20
work page 2001
-
[4]
X. Ren, P. Rinke, C. Joas, M. Scheffler, Random-phase approximation and its applications in compu- tational chemistry and materials science, J. Mater. Sci. 47 (2012) 7447–7471
work page 2012
- [5]
-
[6]
X. Ren, P. Rinke, M. Scheffler, Exploring the random phase approximation: Application to co adsorbed on cu(111), Phys. Rev. B 80 (2009) 045402
work page 2009
-
[7]
J. Harl, L. Schimka, G. Kresse, Assessing the quality of the random phase approximation for lattice constants and atomization energies of solids, Phys. Rev. B 81 (2010) 115126
work page 2010
-
[8]
M. Del Ben, J. Hutter, J. VandeV ondele, Probing the structural and dynamical properties of liquid water with models including non-local electron correlation, J. Chem. Phys. 143 (5) (2015) 054506
work page 2015
-
[9]
Z.-H. Cui, F. Wu, H. Jiang, First-principles study of relative stability of rutile and anatase tio2 using the random phase approximation, Phys. Chem. Chem. Phys. 18 (2016) 29914–29922
work page 2016
-
[10]
J. Hermann, R. A. J. DiStasio, A. Tkatchenko, First-principles models for van der waals interactions in molecules and materials: Concepts, theory, and applications, Chem. Rev. 117 (6) (2017) 4714–4758
work page 2017
-
[11]
P. S. Schmidt, K. S. Thygesen, Benchmark database of transition metal surface and adsorption energies from many-body perturbation theory, J. Phys. Chem. C 122 (8) (2018) 4381–4390
work page 2018
- [12]
- [13]
-
[14]
B. Ramberger, T. Schäfer, G. Kresse, Analytic interatomic forces in the random phase approximation, Phys. Rev. Lett. 118 (2017) 106403. 20
work page 2017
-
[15]
S. Shah, B. Zhang, H. Huang, J. E. Pask, P. Suryanarayana, E. Chow, Many-body electronic correlation energy using krylov subspace linear solvers, in: SC24: International Conference for High Performance Computing, Networking, Storage and Analysis, 2024, pp. 1–15
work page 2024
- [16]
-
[17]
J. D. Talman, W. F. Shadwick, Optimized effective atomic central potential, Phys. Rev. A 14 (1976) 36–40
work page 1976
-
[18]
L. J. Sham, M. Schlüter, Density-functional theory of the energy gap, Phys. Rev. Lett. 51 (1983) 1888– 1891
work page 1983
-
[19]
S. Lehtola, A review on non-relativistic, fully numerical electronic structure calculations on atoms and diatomic molecules, Int. J. Quantum Chem. 119 (19) (2019) e25968
work page 2019
-
[20]
D. R. Hamann, Optimized norm-conserving Vanderbilt pseudopotentials, Phys. Rev. B 88 (8) (2013) 085117
work page 2013
-
[21]
N. Troullier, J. L. Martins, Efficient pseudopotentials for plane-wave calculations, Phys. Rev. B 43 (3) (1991) 1993
work page 1991
-
[22]
M. F. Shojaei, J. E. Pask, A. J. Medford, P. Suryanarayana, Soft and transferable pseudopotentials from multi-objective optimization, Comput. Phys. Comm. 283 (2023) 108594
work page 2023
-
[23]
P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B 50 (24) (1994) 17953
work page 1994
-
[24]
N. A. W. Holzwarth, A. R. Tackett, G. E. Matthews, A projector augmented wave (PAW) code for electronic structure calculations, Part I: atompaw for generating atom-centered functions, Comput. Phys. Comm. 135 (3) (2001) 329–347
work page 2001
-
[25]
V . I. Anisimov, O. Gunnarsson, Density-functional calculation of effective coulomb interactions in metals, Phys. Rev. B 43 (10) (1991) 7570
work page 1991
-
[26]
V . I. Anisimov, J. Zaanen, O. K. Andersen, Band theory and mott insulators: Hubbard U instead of stoner I, Phys. Rev. B 44 (3) (1991) 943
work page 1991
-
[27]
S. Bhowmik, A. J. Medford, P. Suryanarayana, Real-space hubbard-corrected density functional the- ory, J. Chem. Phys. 163 (23)
-
[28]
Lehtola, Assessment of initial guesses for self-consistent field calculations
S. Lehtola, Assessment of initial guesses for self-consistent field calculations. superposition of atomic potentials: Simple yet efficient, J. Chem. Theory Comput. 15 (3) (2019) 1593–1604
work page 2019
-
[29]
S. Lehtola, L. Visscher, E. Engel, Efficient implementation of the superposition of atomic potentials initial guess for electronic structure calculations in gaussian basis sets, J. Chem. Phys. 152 (14) (2020) 144101
work page 2020
-
[30]
Q. Xu, A. Sharma, B. Comer, H. Huang, E. Chow, A. J. Medford, J. E. Pask, P. Suryanarayana, SPARC: Simulation package for ab-initio real-space calculations, SoftwareX 15 (2021) 100709. 21
work page 2021
-
[31]
D. R. Bowler, R. Choudhury, M. J. Gillan, T. Miyazaki, Recent progress with large-scale ab initio calculations: the CONQUEST code, Phys. Status Solidi B 243 (5) (2006) 989–1000
work page 2006
-
[32]
A. García, N. Papior, A. Akhtar, E. Artacho, V . Blum, E. Bosoni, P. Brandimarte, M. Brandbyge, J. I. Cerdá, F. Corsetti, R. Cuadrado, V . Dikan, J. Ferrer, J. Gale, P. García-Fernández, V . M. García-Suárez, S. García, G. Huhs, S. Illera, R. Korytár, P. Koval, I. Lebedeva, L. Lin, P. López-Tarifa, S. G. Mayo, S. Mohr, P. Ordejón, A. Postnikov, Y . Pouill...
work page 2020
-
[33]
J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V . Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V . Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B....
work page 2010
-
[34]
V . Blum, R. Gehrke, F. Hanke, P. Havu, V . Havu, X. Ren, K. Reuter, M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Comm. 180 (11) (2009) 2175–2196
work page 2009
- [35]
-
[36]
V . Wang, N. Xu, J. Liu, G. Tang, W. Geng, Vaspkit: A user-friendly interface facilitating high- throughput computing and analysis using vasp code, Comput. Phys. Comm. 267 (2021) 108033
work page 2021
- [37]
-
[38]
Z. Cao, F. Wang, M. Yang, Coupled-cluster method for open-shell heavy-element systems with spin- orbit coupling, J. Chem. Phys. 146 (13) (2017) 134108
work page 2017
-
[39]
E. R. Davidson, S. A. Hagstrom, S. J. Chakravorty, V . M. Umar, C. F. Fischer, Ground-state correlation energies for two- to ten-electron atomic ions, Phys. Rev. A 44 (1991) 7071–7083
work page 1991
-
[40]
S. J. Chakravorty, S. R. Gwaltney, E. R. Davidson, F. A. Parpia, C. F. Fischer, Ground-state correlation energies for atomic ions with 3 to 18 electrons, Phys. Rev. A 47 (1993) 3649–3670
work page 1993
-
[41]
C. J. Umrigar, X. Gonze, Accurate exchange-correlation potentials and total-energy components for the helium isoelectronic series, Phys. Rev. A 50 (1994) 3827–3837
work page 1994
- [42]
- [43]
-
[44]
S. K. Trivedi, P. Suryanarayana, Spectral finite-element formulation of the optimized effective potential method for atomic structure in the random phase approximation, J. Chem. Phys. 164 (8) (2026) 084108. 22
work page 2026
-
[45]
X. Lei, A. J. Medford, A universal framework for featurization of atomistic systems, J. Phys. Chem. Lett. 13 (34) (2022) 7911–7919
work page 2022
- [46]
-
[47]
Z. Qiao, A. S. Christensen, M. Welborn, F. R. Manby, A. Anandkumar, T. F. Miller III, Informing geometric deep learning with electronic interactions to accelerate quantum chemistry, Proc. Natl. Acad. Sci. U.S.A. 119 (31) (2022) e2205221119
work page 2022
-
[48]
O. ˇCertík, J. E. Pask, J. Vackáˇr, dftatom: A robust and general schrödinger and dirac solver for atomic structure calculations, Comput. Phys. Comm. 184 (7) (2013) 1777–1791
work page 2013
-
[49]
N. A. W. Holzwarth, M. Torrent, J. Charraud, M. Côté, Cubic spline solver for generalized density functional treatments of atoms and generation of atomic datasets for use with exchange-correlation functionals including meta-gga, Phys. Rev. B 105 (12) (2022) 125144
work page 2022
- [50]
-
[51]
S. Lehtola, Meta-gga density functional calculations on atoms with spherically symmetric densities in the finite element formalism, J. Chem. Theory Comput. 19 (9) (2023) 2502–2517
work page 2023
-
[52]
O. ˇCertík, J. E. Pask, I. Fernando, R. Goswami, N. Sukumar, L. A. Collins, G. Manzini, J. Vacká ˇr, High-order finite element method for atomic structure calculations, Comput. Phys. Comm. 297 (2024) 109051
work page 2024
-
[53]
S. Lehtola, Fully numerical calculations on atoms with fractional occupations and range-separated exchange functionals, Phys. Rev. A 101 (1) (2020) 012516
work page 2020
-
[54]
Z. Romanowski, A b-spline finite element solution of the kohn–sham equation for an atom, Modelling Simul. Mater. Sci. Eng. 16 (1) (2007) 015003
work page 2007
-
[55]
Romanowski, Adaptive solver of a kohn–sham equation for an atom, Modelling Simul
Z. Romanowski, Adaptive solver of a kohn–sham equation for an atom, Modelling Simul. Mater. Sci. Eng. 17 (4) (2009) 045001
work page 2009
-
[56]
Lehtola, Fully numerical hartree-fock and density functional calculations
S. Lehtola, Fully numerical hartree-fock and density functional calculations. i. atoms, Int. J. Quantum Chem. 119 (19) (2019) e25945
work page 2019
-
[57]
M. Cinal, Highly accurate numerical solution of hartree–fock equation with pseudospectral method for closed-shell atoms, J. Math. Chem. 58 (8) (2020) 1571–1600
work page 2020
-
[58]
C. F. Fischer, W. Guo, Z. Shen, Spline methods for multiconfiguration hartree–fock calculations, Int. J. Quantum Chem. 42 (4) (1992) 849–867
work page 1992
- [59]
- [60]
-
[61]
M. Hellgren, U. von Barth, Correlation potential in density functional theory at the gwa level: Spherical atoms, Phys. Rev. B 76 (2007) 075107
work page 2007
-
[62]
S. Vacondio, D. Varsano, A. Ruini, A. Ferretti, Numerically precise benchmark of many-body self- energies on spherical atoms, J. Chem. Theory Comput. 18 (6) (2022) 3703–3717
work page 2022
-
[63]
E. Trushin, S. Fauser, A. Mölkner, J. Erhard, A. Görling, Accurate correlation potentials from the self-consistent random phase approximation, Phys. Rev. Lett. 134 (2025) 016402
work page 2025
-
[64]
S. Bhowmik, J. E. Pask, A. J. Medford, P. Suryanarayana, Spectral scheme for atomic structure calcu- lations in density functional theory, Comput. Phys. Commun. 308 (2025) 109448
work page 2025
- [65]
-
[66]
M. J. T. Oliveira, F. Nogueira, Generating relativistic pseudo-potentials with explicit incorporation of semi-core states using ape, the atomic pseudo-potentials engine, Comput. Phys. Commun. 178 (7) (2008) 524–534
work page 2008
-
[67]
A. P. Bartók, J. R. Yates, Ultrasoft pseudopotentials with kinetic energy density support: Implementing the tran-blaha potential, Phys. Rev. B 99 (2019) 235103
work page 2019
-
[68]
J. Yang, L. Z. Tan, A. M. Rappe, Hybrid functional pseudopotentials, Phys. Rev. B 97 (2018) 085130
work page 2018
-
[69]
Y . Yang, G. Prokopiou, T. Qiu, A. M. Schankler, A. M. Rappe, L. Kronik, R. A. DiStasio, Range- separated hybrid functional pseudopotentials, Phys. Rev. B 108 (2023) 165142
work page 2023
-
[70]
J. P. Perdew, K. Burke, M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77 (18) (1996) 3865
work page 1996
- [71]
- [72]
-
[73]
A. Görling, M. Levy, Exact kohn-sham scheme based on perturbation theory, Phys. Rev. A 50 (1994) 196–204
work page 1994
- [74]
-
[75]
Kleinman, Relativistic norm-conserving pseudopotential, Phys
L. Kleinman, Relativistic norm-conserving pseudopotential, Phys. Rev. B 21 (6) (1980) 2630
work page 1980
-
[76]
A. T. Patera, A spectral element method for fluid dynamics: Laminar flow in a channel expansion, J. Comput. Phys. 54 (3) (1984) 468–488
work page 1984
-
[77]
S. H. V osko, L. Wilk, M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys. 58 (8) (1980) 1200–1211
work page 1980
-
[78]
J. P. Perdew, A. Zunger, Self-interaction correction to density-functional approximations for many- electron systems, Phys. Rev. B 23 (10) (1981) 5048. 24
work page 1981
-
[79]
J. P. Perdew, Y . Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B 45 (23) (1992) 13244
work page 1992
-
[80]
J. Sun, A. Ruzsinszky, J. P. Perdew, Strongly constrained and appropriately normed semilocal density functional, Phys. Rev. Lett. 115 (3) (2015) 036402
work page 2015
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.