Direct Simulation of LiNi0.8Mn0.1Co0.1O2 Transport Properties Using an Efficient and Accurate Machine Learning Potential
Pith reviewed 2026-05-20 04:25 UTC · model grok-4.3
The pith
A machine learning potential built from limited DFT data enables direct large-scale simulation of lithium self-diffusion in NMC811.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
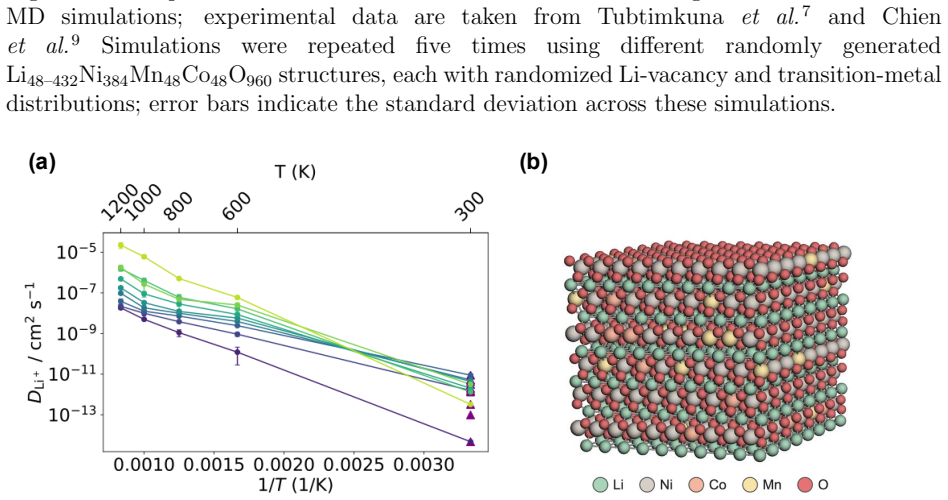
The authors show that a machine learning potential for LiNi0.8Mn0.1Co0.1O2, constructed by fine-tuning a MACE foundation model and refining it through active learning on a near-ground-state dataset with a limited number of DFT calculations, supports molecular dynamics trajectories that yield lithium diffusion coefficients while preserving DFT-level accuracy at time and length scales inaccessible to direct density functional theory.
What carries the argument
The machine learning potential for NMC811 trained via active learning on DFT data, which serves as a fast surrogate for the potential energy surface in molecular dynamics of lithium ion transport.
If this is right
- Lithium self-diffusion coefficients in NMC811 become directly computable from long, large molecular dynamics trajectories.
- Transport mechanisms can be examined at scales where collective effects and realistic defect concentrations appear.
- The same potential can be reused for repeated simulations under varied temperatures or compositions without new DFT runs.
Where Pith is reading between the lines
- The method could be transferred to related layered oxides to map how transition-metal ratios change diffusion rates.
- Longer trajectories made possible by the potential might reveal rare events such as lithium trapping near defects.
- Direct access to diffusion data at experimental length scales could tighten the link between atomistic models and measured rate capability.
Load-bearing premise
The errors remaining in the trained machine learning potential do not meaningfully alter the lithium diffusion pathways or energy barriers that appear in the molecular dynamics runs.
What would settle it
Running short DFT-based molecular dynamics on small cells and finding that the lithium diffusion coefficients or hop barriers differ substantially from those obtained with the machine learning potential on the same cells.
Figures





read the original abstract
The rate capability of layered lithium nickel manganese cobalt oxide (NMC) cathode materials plays a decisive role in high-power applications such as fast charging, necessitating a detailed understanding of lithium-ion diffusion. However, the mechanisms governing lithium-ion transport in NMC remain insufficiently understood, both experimentally and computationally. In this study, we employ an advanced and efficient machine learning potential (MLP) to simulate lithium self-diffusion in LiNi0.8Mn0.1Co0.1O2 (NMC811), enabling direct large-scale molecular dynamics (MD) simulations. The workflow integrates a fine-tuned MACE (Message Passing Atomic Cluster Expansion) foundation model as a structural generator and leverages an active learning strategy applied to a near-ground-state dataset. This approach enables the construction of a reliable MLP for NMC811 in a data-efficient manner using a limited number of density functional theory (DFT) reference calculations. Based on this potential, we performed MD simulations to predict lithium diffusion coefficients. The MLP-based simulations preserve the accuracy of DFT while overcoming its time and length scale limitations, thereby allowing direct simulation of lithium self-diffusion in NMC811.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops a machine learning potential (MLP) for LiNi0.8Mn0.1Co0.1O2 (NMC811) by fine-tuning a MACE foundation model and applying active learning to a near-ground-state dataset using a limited number of DFT calculations. This MLP is then employed in large-scale molecular dynamics simulations to compute lithium self-diffusion coefficients, with the central claim that the approach preserves DFT accuracy while overcoming the time- and length-scale limitations of direct DFT-based simulations.
Significance. If the MLP accurately reproduces DFT-level barriers and pathways, the work would enable direct, large-scale simulation of activated Li transport in high-Ni layered cathodes, providing mechanistic insight into rate-limiting processes relevant to fast-charging battery applications.
major comments (2)
- [Methods section describing the active-learning protocol] The active learning workflow is described as being applied exclusively to a near-ground-state dataset. Because Li diffusion in NMC811 is an activated process whose rate depends exponentially on barrier height, configurations near octahedral-to-tetrahedral saddle points or those involving local transition-metal disorder lie outside this manifold. Without reported validation of force or energy errors on such transition-state structures (or direct comparison of MLP vs. DFT barrier heights), it remains unclear whether the extracted MD diffusion coefficients are free of systematic bias.
- [Results section on MD-derived transport properties] The results section reports lithium diffusion coefficients from MLP-MD but provides neither error bars on the coefficients nor quantitative benchmarks against experimental tracer diffusion data or prior ab initio MD studies. Such comparisons are required to substantiate the claim that the MLP 'preserves the accuracy of DFT'.
minor comments (1)
- [Abstract] The abstract asserts that the MLP 'preserves the accuracy of DFT' without accompanying quantitative metrics (e.g., force MAE on validation sets or barrier errors); this phrasing should be qualified or supported by explicit numbers in the main text.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed review. The comments raise important points about validation of the MLP for activated diffusion processes and the need for quantitative benchmarks. We have addressed both concerns by planning additional analyses and comparisons that will be incorporated into the revised manuscript.
read point-by-point responses
-
Referee: [Methods section describing the active-learning protocol] The active learning workflow is described as being applied exclusively to a near-ground-state dataset. Because Li diffusion in NMC811 is an activated process whose rate depends exponentially on barrier height, configurations near octahedral-to-tetrahedral saddle points or those involving local transition-metal disorder lie outside this manifold. Without reported validation of force or energy errors on such transition-state structures (or direct comparison of MLP vs. DFT barrier heights), it remains unclear whether the extracted MD diffusion coefficients are free of systematic bias.
Authors: We appreciate the referee's emphasis on the need to validate the MLP specifically for transition-state configurations, given the exponential sensitivity of diffusion rates to barrier heights. The active-learning protocol in the original manuscript was deliberately restricted to near-ground-state structures to achieve data efficiency with a limited DFT budget. To directly address this concern, the revised manuscript will include additional DFT reference calculations on representative saddle-point and disordered configurations (sampled via short exploratory DFT-MD and NEB paths). We will report force and energy errors on these structures and provide a side-by-side comparison of MLP-predicted versus DFT-computed Li diffusion barriers for the dominant octahedral-to-tetrahedral hops. These new results will be presented in an expanded Methods section and a supplementary figure, demonstrating that the MLP reproduces the relevant barriers without introducing systematic bias. revision: yes
-
Referee: [Results section on MD-derived transport properties] The results section reports lithium diffusion coefficients from MLP-MD but provides neither error bars on the coefficients nor quantitative benchmarks against experimental tracer diffusion data or prior ab initio MD studies. Such comparisons are required to substantiate the claim that the MLP 'preserves the accuracy of DFT'.
Authors: We agree that error estimates and external benchmarks are necessary to strengthen the accuracy claim. In the revised manuscript we will add statistical error bars to all reported lithium self-diffusion coefficients, derived from the standard error across at least five independent, long MD trajectories. We will also insert a new comparison table (or figure) that quantitatively benchmarks the MLP-MD diffusivities against available experimental tracer-diffusion data for NMC811 and against prior ab initio MD results for related NMC compositions. These additions will be placed in the Results section and will allow readers to directly assess how well the MLP-MD values align with both experiment and direct DFT simulations. revision: yes
Circularity Check
No significant circularity detected in derivation chain
full rationale
The paper's central workflow begins with external DFT reference calculations, applies active learning to train an MLP on a near-ground-state dataset, and then runs MD simulations to extract lithium self-diffusion coefficients. This chain depends on independent quantum-mechanical data and standard dynamical propagation rather than any self-referential fitting, parameter renaming, or load-bearing self-citation. No equation or step reduces by construction to a quantity already determined inside the paper; the reported transport properties are genuine predictions from the trained potential and the dynamics.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption The MACE foundation model can be fine-tuned to NMC811 chemistry with a small number of DFT calculations while retaining transferability to diffusion events.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The workflow integrates a fine-tuned MACE foundation model as a structural generator and leverages an active learning strategy applied to a near-ground-state dataset... NEB calculations... MD simulations... Einstein relation for diffusion coefficient
-
IndisputableMonolith/Foundation/ArrowOfTime.leanentropy_from_berry unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
lithium self-diffusion coefficients... Arrhenius extrapolation
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Noh, H.-J.; Youn, S.; Yoon, C. S.; Sun, Y.-K. Comparison of the structural and electro- chemical properties of layered Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) 17 cathode material for lithium-ion batteries.J. Power Sources2013,233, 121–130
-
[2]
Hierarchical nickel valence gradient stabilizes high-nickel content layered cathode materials.Nat
Lin, R.; Bak, S.-M.; Shin, Y.; Zhang, R.; Wang, C.; Kisslinger, K.; Ge, M.; Huang, X.; Shadike, Z.; Pattammattel, A. Hierarchical nickel valence gradient stabilizes high-nickel content layered cathode materials.Nat. Commun.2021,12, 2350
work page 2021
-
[3]
Li, W.; Erickson, E. M.; Manthiram, A. High-nickel layered oxide cathodes for lithium- based automotive batteries.Nat. Energy2020,5, 26–34
-
[4]
Kasnatscheew, J.; Evertz, M.; Kloepsch, R.; Streipert, B.; Wagner, R.; Cekic Laskovic, I.; Winter, M. Learning from Electrochemical Data: Simple Evaluation and Classification of LiMO2-type-based Positive Electrodes for Li-Ion Batteries.Energy Technol.2017,5, 1670–1679
work page 2017
-
[5]
McClelland, I.; Booth, S. G.; Anthonisamy, N. N.; Middlemiss, L. A.; P´ erez, G. E.; Cussen, E. J.; Baker, P. J.; Cussen, S. A. Direct Observation of Dynamic Lithium Diffusion Behavior in Nickel-Rich, LiNi 0.8 Mn0.1 Co0.1 O2 (NMC811) Cathodes Using OperandoMuon Spectroscopy.Chem. Mater.2023,35, 4149–4158
work page 2023
-
[6]
C. McNulty, R.; Hampson, E.; N. Cutler, L.; P. Grey, C.; M. Dose, W.; R. Johnson, L. Understanding the limits of Li-NMC811 half-cells.J. Mater. Chem. A2023,11, 18302– 18312
-
[7]
Tubtimkuna, S.; Phattharasupakun, N.; Bunyanidhi, P.; Sawangphruk, M. Diffusion of Zirconium (IV) Ions from Coated Thick Zirconium Oxide Shell to the Bulk Structure of Ni-Rich NMC811 Cathode Leading to High-Performance 18650 Cylindrical Li-Ion Batteries.Adv. Mater. Technol.2022,7, 2200436
work page 2022
-
[8]
M¨ arker, K.; Reeves, P. J.; Xu, C.; Griffith, K. J.; Grey, C. P. Evolution of Structure and Lithium Dynamics in LiNi0.8 Mn0.1 Co0.1 O2 (NMC811) Cathodes during Electrochemical Cycling.Chem. Mater.2019,31, 2545–2554. 18
work page 2019
-
[9]
Chien, Y.-C.; Liu, H.; Menon, A. S.; Brant, W. R.; Brandell, D.; Lacey, M. J. Rapid determination of solid-state diffusion coefficients in Li-based batteries via intermittent current interruption method.Nat. Commun.2023,14, 2289
work page 2023
-
[10]
Van der Ven, A.; Ceder, G.; Asta, M.; Tepesch, P. D. First-principles theory of ionic diffusion with nondilute carriers.Phys. Rev. B2001,64, 184307
-
[11]
Ven, A. V. d.; Ceder, G. Lithium Diffusion in Layered LixCoO2.Electrochem. Solid-State Lett.2000,3, 301
work page 2000
-
[12]
Lithium diffusion mechanisms in layered intercalation com- pounds.J
Van der Ven, A.; Ceder, G. Lithium diffusion mechanisms in layered intercalation com- pounds.J. Power Sources2001,97-98, 529–531
-
[13]
Hoang, K.; Johannes, M. Defect Physics and Chemistry in Layered Mixed Transition Metal Oxide Cathode Materials: (Ni,Co,Mn) vs (Ni,Co,Al).Chem. Mater.2016,28, 1325–1334
work page 2016
-
[14]
Jaberi, A.; Trudeau, M. L.; Song, J.; Gauvin, R. Study of Lithium Transport in NMC Layered Oxide Cathode Material Using Multiscale Computational Approach.ACS Appl. Energy Mater.2024,7, 7724–7736
work page 2024
-
[15]
S.; Markovsky, B.; Aurbach, D.; Major, D
Dixit, M.; Kosa, M.; Lavi, O. S.; Markovsky, B.; Aurbach, D.; Major, D. T. Thermody- namic and kinetic studies of LiNi 0.5 Co 0.2 Mn 0.3 O 2 as a positive electrode material for Li-ion batteries using first principles.Phys. Chem. Chem. Phys.2016,18, 6799–6812
work page 2016
-
[16]
Accelerated Atomistic Modeling of Solid-State Battery Materials With Machine Learning.Front
Guo, H.; Wang, Q.; Stuke, A.; Urban, A.; Artrith, N. Accelerated Atomistic Modeling of Solid-State Battery Materials With Machine Learning.Front. Energy Res.2021,9
work page 2021
-
[17]
Generalized Neural-Network Representation of High- Dimensional Potential-Energy Surfaces.Phys
Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High- Dimensional Potential-Energy Surfaces.Phys. Rev. Lett.2007,98, 146401
work page 2007
-
[18]
Bart´ ok, A. P.; Kondor, R.; Cs´ anyi, G. On representing chemical environments.Phys. Rev. B2013,87, 184115. 19
-
[19]
Artrith, N.; Urban, A. An implementation of artificial neural-network potentials for atomistic materials simulations: Performance for TiO2.Comput. Mater. Sci.2016,114, 135–150
work page 2016
-
[20]
Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics.Phys
Zhang, L.; Han, J.; Wang, H.; Car, R.; E, W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics.Phys. Rev. Lett.2018,120, 143001
work page 2018
-
[21]
Batatia, I.; Batzner, S.; Kov´ acs, D. P.; Musaelian, A.; Simm, G. N. C.; Drautz, R.; Ort- ner, C.; Kozinsky, B.; Cs´ anyi, G. The Design Space of E(3)-Equivariant Atom-Centered Interatomic Potentials. 2022;http://arxiv.org/abs/2205.06643
-
[22]
P.; Simm, G.; Ortner, C.; Csanyi, G
Batatia, I.; Kovacs, D. P.; Simm, G.; Ortner, C.; Csanyi, G. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields.Adv. Neu. Inf. Pro. Sys.2022,35, 11423–11436
work page 2022
-
[23]
Kresse, G.; Furthm¨ uller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set.Comput. Mater. Sci.1996,6, 15–50
work page 1996
-
[24]
Kresse, G.; Furthm¨ uller, J. Efficient iterative schemes for ab initio total-energy calcula- tions using a plane-wave basis set.Phys. Rev. B1996,54, 11169–11186
-
[25]
Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple.Phys. Rev. Lett.1996,77, 3865–3868
work page 1996
-
[26]
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu.J. Chem. Phys.2010,132, 154104
work page 2010
-
[27]
Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems.Int
Himmetoglu, B.; Floris, A.; De Gironcoli, S.; Cococcioni, M. Hubbard-corrected DFT energy functionals: The LDA+U description of correlated systems.Int. J. Qua. Chem. 2014,114, 14–49. 20
work page 2014
-
[28]
Zhou, F.; Cococcioni, M.; Marianetti, C. A.; Morgan, D.; Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA + U.Phys. Rev. B2004,70, 235121
-
[29]
C.; Bare˜ no, J.; Yan, J.; Chen, G.; Hauser, A.; Croy, J
Garcia, J. C.; Bare˜ no, J.; Yan, J.; Chen, G.; Hauser, A.; Croy, J. R.; Iddir, H. Surface Structure, Morphology, and Stability of Li(Ni 1/3 Mn1/3 Co1/3 )O2 Cathode Material.J. Phys. Chem. C2017,121, 8290–8299
-
[30]
Schipper, F.; Dixit, M.; Kovacheva, D.; Talianker, M.; Haik, O.; Grinblat, J.; Erick- son, E. M.; Ghanty, C.; Major, D. T.; Markovsky, B. Stabilizing nickel-rich layered cathode materials by a high-charge cation doping strategy: zirconium-doped LiNi 0.6 Co 0.2 Mn 0.2 O 2.J. Mater. Chem. A2016,4, 16073–16084
-
[31]
Artrith, N.; Urban, A.; Ceder, G. Constructing first-principles phase diagrams of amor- phous LixSi using machine-learning-assisted sampling with an evolutionary algorithm. J. Chem. Phys.2018,148
work page 2018
-
[33]
Kong, L.; Li, J.; Sun, L.; Yang, H.; Hao, H.; Chen, C.; Artrith, N.; Torres, J. A. G.; Lu, Z.; Zhou, Y. Overcoming the Size Limit of First Principles Molecular Dynamics Simulations with an In-Distribution Substructure Embedding Active Learner.2023,
work page 2023
-
[34]
Zhang, Y.; Wang, H.; Chen, W.; Zeng, J.; Zhang, L.; Wang, H.; E, W. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models.Com. Phys. Commun.2020,253, 107206
work page 2020
-
[35]
Andrade, M. F. C.; Ko, H.-Y.; Zhang, L.; Car, R.; Selloni, A. Free energy of proton transfer at the water–TiO 2 interface from ab initio deep potential molecular dynamics. Chem. Sci.2020,11, 2335–2341. 21
work page 2020
-
[36]
Chen, J.; Fang, M.; Wu, Q.; Tang, S.; Zheng, J.; Wei, C.; Cao, X.; Shi, Y.; Xu, N.; He, Y. Insights into the Atomic Mechanism of Lithium-Ion Diffusion in Li 6PS5Cl via a Machine Learning Potential.Chem. Mater.2025,37, 591–599
work page 2025
-
[37]
Jia, W.; Wang, H.; Chen, M.; Lu, D.; Lin, L.; Car, R.; Weinan, E.; Zhang, L. Pushing the Limit of Molecular Dynamics with Ab Initio Accuracy to 100 Million Atoms with Machine Learning. Proc. Int. Conf. High Perform. Comput. Netw. Storage Anal. 2020; pp 1–14
work page 2020
-
[38]
Artrith, N.; Urban, A.; Ceder, G. Efficient and accurate machine-learning interpolation of atomic energies in compositions with many species.Phys. Rev. B2017,96, 014112. 22 Supporting Information Direct Simulation of LiNi 0.8Mn0.1Co0.1O2 Transport Properties Using an Efficient and Accurate Machine Learning Potential Jian He, Constantijn H. J. A. van de We...
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.