Additive binding energies in asphalt on a quantum processor via quantum-selected configuration interaction (QSCI)
Pith reviewed 2026-06-29 16:43 UTC · model grok-4.3
The pith
A quantum processor computes the binding energy of an asphalt hydrogen-bond model exactly matching the classical active-space reference.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
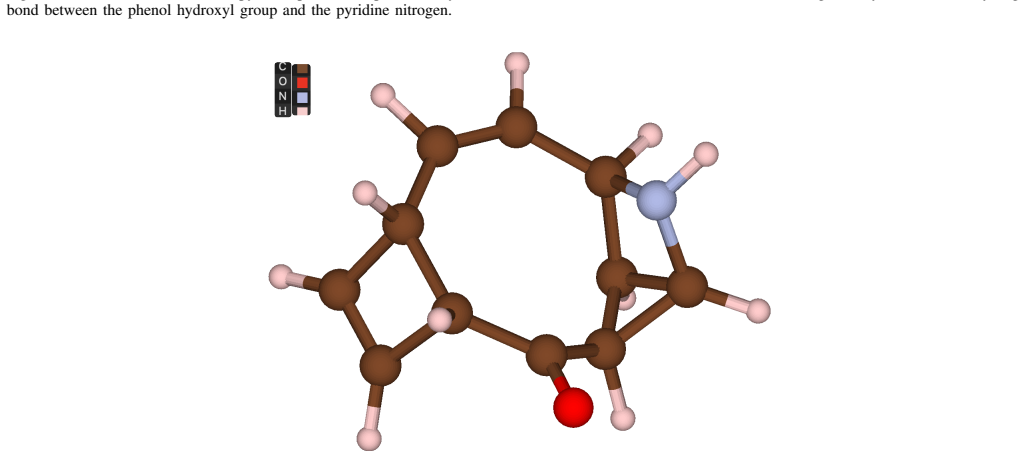
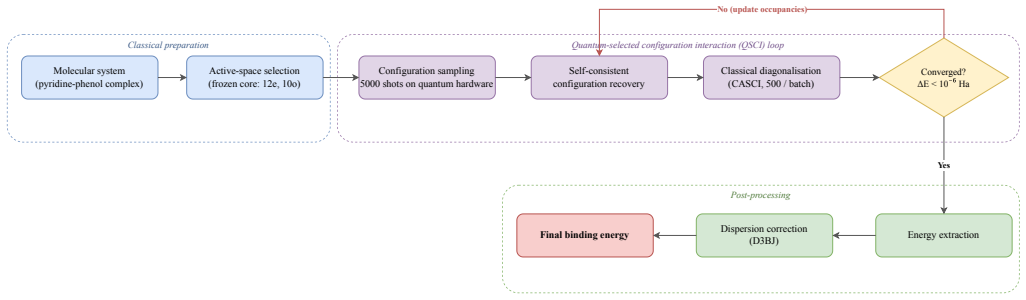
In the QuantumPave workflow, machine-learning interatomic potentials first optimize the geometry of the 24-atom pyridine-phenol hydrogen-bonded complex; quantum-selected configuration interaction (QSCI, or SQD) then samples configurations on the 54-qubit IQM Emerald processor in a (10e,10o) active space and classical resources complete the diagonalization. On hardware this reproduces the active-space CASCI binding energy of -3.52 kcal/mol exactly, capturing static correlation within the chosen orbitals while underbinding relative to a counterpoise-corrected CCSD(T) benchmark of -8.5 to -9.5 kcal/mol.
What carries the argument
Quantum-selected configuration interaction (QSCI), also called sample-based quantum diagonalization (SQD), in which the quantum processor samples the dominant electronic configurations and classical high-performance computing resources perform the subsequent diagonalization.
If this is right
- Device noise broadens sampling across the active space, eliminating any need for zero-noise extrapolation.
- The active-space result captures static correlation but remains lower than full CCSD(T) benchmarks and experimental enthalpy values once thermal and solvent effects are considered.
- The workflow integrates classical machine-learning potentials for geometry with quantum computation for the electronic structure problem.
Where Pith is reading between the lines
- Scaling the active space or model size could allow the same hardware sampling approach to treat more complex asphalt components.
- The method opens a route to quantum-assisted calculations on other hydrogen-bonded materials systems where static correlation matters.
- Adding perturbative or density-functional corrections on top of the active-space QSCI result might close the gap to higher-level benchmarks without increasing quantum resources.
Load-bearing premise
The 24-atom pyridine-phenol complex and chosen (10e,10o) active space are representative enough of the hydrogen bonding that governs oxidative ageing in real asphalt binder.
What would settle it
Running the same (10e,10o) active-space calculation on the quantum processor and obtaining a binding energy that deviates from the classical CASCI reference would falsify the claim of exact reproduction on current hardware.
Figures

read the original abstract
Quantum-centric supercomputing (in which a quantum processor samples the dominant electronic configurations and classical high-performance computing resources perform the diagonalisation) is emerging as a practical route to correlated electronic-structure calculations. We present QuantumPave, a hybrid quantum-classical workflow for computing additive binding energies in asphalt binder, a quantity central to the oxidative ageing of road infrastructure. Using a 24-atom pyridine-phenol hydrogen-bonded complex as a representative model, we couple machine-learning interatomic potentials (ORB v3) for geometry optimisation with quantum-selected configuration interaction (QSCI), also referred to as sample based quantum diagonalisation (SQD), in a (10e, 10o) active space run on the 54-qubit IQM Emerald processor. On hardware, SQD reproduces the active-space CASCI reference exactly, giving a binding energy of -3.52 kcal/mol (-0.153 eV); the device noise broadens the sampling to span the active space, so no zero-noise extrapolation is required. This active-space value captures the static correlation within the chosen orbitals and underbinds the full hydrogen bond: a counterpoise-corrected CCSD(T) benchmark gives -8.5 to -9.5 kcal/mol, while the calorimetric enthalpy of about -6.25 kcal/mol is consistent with this once zero-point, thermal, and solvent contributions are included. We show that chemically meaningful binding energies for an industrially relevant materials problem are attainable on current quantum hardware within a quantum-centric supercomputing workflow.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents QuantumPave, a hybrid quantum-classical workflow that couples machine-learning interatomic potentials (ORB v3) for geometry optimization with quantum-selected configuration interaction (QSCI/SQD) on a (10e,10o) active space executed on the 54-qubit IQM Emerald processor. Using a 24-atom pyridine-phenol hydrogen-bonded complex as a model for hydrogen bonding in asphalt binder, the hardware SQD calculation exactly reproduces the CASCI reference, producing a binding energy of -3.52 kcal/mol (-0.153 eV). This value is compared to counterpoise-corrected CCSD(T) benchmarks (-8.5 to -9.5 kcal/mol) and experimental calorimetric enthalpy (~-6.25 kcal/mol), with the claim that chemically meaningful binding energies for an industrially relevant materials problem are attainable on current quantum hardware within a quantum-centric supercomputing workflow.
Significance. If the central assumptions hold, the work provides a concrete demonstration that current quantum processors can deliver exact active-space results for a materials-relevant binding-energy problem without zero-noise extrapolation, advancing quantum-centric supercomputing workflows that combine ML potentials with quantum sampling. The exact hardware-CASCI match is a clear technical strength.
major comments (2)
- [Abstract] Abstract: The 24-atom pyridine-phenol complex and (10e,10o) active space are presented as a 'representative model' for the hydrogen bonding that governs oxidative ageing in asphalt, yet no supporting evidence (binding-energy convergence with larger fragments, structural metrics, or direct comparison to experimental asphalt data) is supplied. This assumption is load-bearing for the central claim, especially since the reported active-space binding energy underbinds the CCSD(T) benchmark by a factor of ~2.5.
- [Abstract] Abstract: No test is provided showing that enlarging the active space or model size to recover the missing dynamic correlation (needed to approach the CCSD(T) or experimental values) would remain feasible on the available hardware, leaving open whether the demonstrated workflow scales to chemically accurate asphalt binding energies.
minor comments (1)
- The abstract would benefit from an explicit statement of the active-space limitations and the precise definition of 'additive binding energies' used in the workflow.
Simulated Author's Rebuttal
We thank the referee for the constructive comments. We respond point-by-point below, agreeing where the manuscript requires clarification or additional discussion.
read point-by-point responses
-
Referee: [Abstract] Abstract: The 24-atom pyridine-phenol complex and (10e,10o) active space are presented as a 'representative model' for the hydrogen bonding that governs oxidative ageing in asphalt, yet no supporting evidence (binding-energy convergence with larger fragments, structural metrics, or direct comparison to experimental asphalt data) is supplied. This assumption is load-bearing for the central claim, especially since the reported active-space binding energy underbinds the CCSD(T) benchmark by a factor of ~2.5.
Authors: The pyridine-phenol complex was selected because pyridine and phenol fragments appear in asphalt oxidation chemistry and form a clean hydrogen-bonded dimer amenable to active-space treatment. We acknowledge that the manuscript supplies no convergence data with larger fragments, no structural metrics against asphalt models, and no direct experimental asphalt comparison, so the representativeness claim rests on chemical analogy alone. The underbinding relative to CCSD(T) is already stated in the text as a direct consequence of restricting to static correlation in (10e,10o). We will revise the abstract and introduction to describe the system explicitly as an illustrative model chosen for computational tractability rather than a validated proxy, thereby reducing the load-bearing weight of the assumption. revision: yes
-
Referee: [Abstract] Abstract: No test is provided showing that enlarging the active space or model size to recover the missing dynamic correlation (needed to approach the CCSD(T) or experimental values) would remain feasible on the available hardware, leaving open whether the demonstrated workflow scales to chemically accurate asphalt binding energies.
Authors: We agree that the manuscript contains no explicit test of larger active spaces or molecular fragments. The (10e,10o) space was the largest for which exact SQD sampling and diagonalization could be performed on the 54-qubit IQM Emerald device with the chosen qubit mapping. The quantum-centric workflow is constructed so that larger problems map onto additional qubits or improved sampling; however, demonstrating this scaling on current hardware lies outside the scope of the present demonstration. We will add a concise paragraph in the discussion section outlining the qubit and sampling requirements for recovering dynamic correlation and the expected path to chemical accuracy with next-generation processors. revision: yes
Circularity Check
No circularity: direct hardware computation of active-space energy compared to independent classical benchmarks
full rationale
The paper computes the binding energy by running QSCI/SQD on the IQM Emerald processor for the fixed (10e,10o) active space of the 24-atom model, obtaining a value that exactly reproduces the classical CASCI reference within the active space. This result is then compared to separate CCSD(T) calculations and experimental calorimetry; neither the active-space energy nor the binding energy is obtained by fitting parameters to the target quantity or by any self-referential definition. The workflow (ML geometry optimization + quantum sampling + classical diagonalization) contains no load-bearing self-citation chain or ansatz that reduces the reported number to an input by construction. The choice of model system and active space is an assumption whose representativeness can be debated, but that choice does not create a circular derivation.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Chemical composition of asphalt as related to asphalt durability,
J. C. Petersen, “Chemical composition of asphalt as related to asphalt durability,”Developments in Petroleum Science, vol. 40, pp. 363–399, 2000
2000
-
[2]
Review of interfacial adhesion between asphalt and aggregate based on molecular dynamics,
J. yun Xu, B. Ma, W. jie Mao, W. Si, and X. Wang, “Review of interfacial adhesion between asphalt and aggregate based on molecular dynamics,”Construction and Building Materials, vol. 362, p. 129642, 2023. [Online]. Available: https://www.sciencedirect.com/ science/article/pii/S0950061822032986
2023
-
[3]
Evolution of model com- pounds and functional group compositions for molecular dynamics simulations of aged asphalt binder,
E. A. O’Rear, L. Huang, and M. Zaman, “Evolution of model com- pounds and functional group compositions for molecular dynamics simulations of aged asphalt binder,”Molecules, vol. 30, no. 22, p. 4476, 2025
2025
-
[4]
Study on the effect of aging on physical properties of asphalt binder from a microscale perspective,
X. Qu, Q. Liu, M. Guo, D. Wang, and M. Oeser, “Study on the effect of aging on physical properties of asphalt binder from a microscale perspective,”Construction and Building Materials, vol. 187, pp. 718– 729, 2018
2018
-
[5]
Quantum computational chemistry,
S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin, and X. Yuan, “Quantum computational chemistry,”Rev. Mod. Phys., vol. 92, no. 1, p. 015003, 2020
2020
-
[6]
Quantum algorithms for quantum chemistry and quantum materials science,
B. Bauer, S. Bravyi, M. Motta, and G. K.-L. Chan, “Quantum algorithms for quantum chemistry and quantum materials science,”Chemical Re- views, vol. 120, no. 22, pp. 12 685–12 717, 2020
2020
-
[7]
Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer,
J. Robledo-Moreno, M. Motta, H. Haas, A. Javadi-Abhari, P. Jurcevic, W. Kirby, S. Martiel, K. Sharma, S. Sharma, T. Shirakawa, I. Sitdikov, R.-Y . Sun, K. J. Sung, M. Takita, M. C. Tran, S. Yunoki, and A. Mezzacapo, “Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer,”Science Advances, vol. 11, p. eadu9991, 2025. [Onlin...
-
[8]
Simulated quantum computation of molecular energies,
A. Aspuru-Guzik, A. D. Dutoi, P. J. Love, and M. Head-Gordon, “Simulated quantum computation of molecular energies,”Science, vol. 309, no. 5741, pp. 1704–1707, 2005
2005
-
[9]
Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,
A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow, and J. M. Gambetta, “Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets,”Nature, vol. 549, no. 7671, pp. 242–246, 2017
2017
-
[10]
Simulation of a diels–alder reaction on a quantum computer,
I. Liepuoniute, M. Motta, T. Pellegrini, J. E. Rice, T. P. Gujarati, S. Gil, and G. O. Jones, “Simulation of a diels–alder reaction on a quantum computer,”Physical Chemistry Chemical Physics, vol. 26, pp. 25 181– 25 191, 2024
2024
-
[11]
Beyond nisq: The megaquop machine,
J. Preskill, “Beyond nisq: The megaquop machine,”arXiv preprint arXiv:2502.17368, 2025
-
[12]
Pyridine interactions with phenolic groups in water: evi- dence for hydrogen bonding and hydrophobic association,
A. E. Pekary, “Pyridine interactions with phenolic groups in water: evi- dence for hydrogen bonding and hydrophobic association,”Biophysical Chemistry, vol. 7, no. 4, pp. 325–338, 1978
1978
-
[13]
Reference architecture of a quantum-centric supercomputer,
S. Seelam, J. M. Chow, A. C ´orcoles, S. Sheldon, T. Mittal, A. Kandala, S. Dague, I. Hincks, H. Horii, B. Johnson, M. Le, H. Jamjoom, and J. M. Gambetta, “Reference architecture of a quantum-centric supercomputer,” arXiv preprint arXiv:2603.10970, 2026
-
[14]
K. M. Merz Jr., A. Shajan, D. Kaliakin, F. Lianget al., “Crossing the 12,000-atom barrier with heterogeneous quantum-classical supercom- puting: quantum chemistry of protein-ligand complexes,”arXiv preprint arXiv:2605.01138, 2026
work page internal anchor Pith review Pith/arXiv arXiv 2026
-
[15]
Quantum-accelerated supercomputing atomistic simulations for corrosion inhibition,
K. Elgammal and M. Maußner, “Quantum-accelerated supercomputing atomistic simulations for corrosion inhibition,” inISC High Performance 2025 Research Paper Proceedings (40th International Conference), 2025, pp. 1–10, arXiv:2412.00951
-
[16]
K. Kannoet al., “Quantum-selected configuration interaction: Classical diagonalization of Hamiltonians in subspaces selected by quantum computers,”arXiv:2302.11320, 2023. [Online]. Available: https: //arxiv.org/abs/2302.11320
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[17]
S. Barison, J. Robledo Moreno, and M. Motta, “Quantum-centric computation of molecular excited states with extended sample- based quantum diagonalization,”Quantum Science and Technology, vol. 10, no. 2, p. 025034, Feb. 2025. [Online]. Available: http: //dx.doi.org/10.1088/2058-9565/adb781
-
[18]
Quantum-centric algorithm for sample-based krylov diagonalization,
J. Yu, J. R. Moreno, J. T. Iosue, L. Bertels, D. Claudino, B. Fuller, P. Groszkowski, T. S. Humble, P. Jurcevic, W. Kirby, T. A. Maier, M. Motta, B. Pokharel, A. Seif, A. Shehata, K. J. Sung, M. C. Tran, V . Tripathi, A. Mezzacapo, and K. Sharma, “Quantum-centric algorithm for sample-based krylov diagonalization,”arXiv preprint arXiv:2501.09702, 2025
-
[19]
S. Shivpuje, T. P. Gujaratiet al., “Sample-based quantum diagonaliza- tion methods for modeling the photochemistry of diazirine and diazo compounds,”arXiv preprint arXiv:2510.00484, 2025
-
[20]
Hybrid quantum algorithms for computational chemistry: Application to the pyridine-li ion complex,
F. Ghasemi, Y . Kang, Y . Kawashima, and K. Moon, “Hybrid quantum algorithms for computational chemistry: Application to the pyridine-li ion complex,”arXiv preprint arXiv:2601.10002, 2026
-
[21]
Orb: A fast, scalable neural network potential,
M. Neumann, J. Gin, B. Rhodes, S. Bennett, Z. Li, H. Choubisa, A. Hussey, and J. Godwin, “Orb: A fast, scalable neural network potential,” 2024. [Online]. Available: https://arxiv.org/abs/2410.22570
-
[22]
Orb-v3: atomistic simulation at scale,
B. Rhodes, S. Vandenhaute, V . ˇSimkuset al., “Orb-v3: atomistic simulation at scale,”arXiv preprint arXiv:2504.06231, 2025
-
[23]
J. H ¨anseroth, A. Fl ¨otottoet al., “Fine-tuning unifies foundational machine-learned interatomic potential architectures at ab initio accu- racy,”arXiv preprint arXiv:2511.05337, 2025
-
[24]
The calculation of small molecular inter- actions by the differences of separate total energies. some procedures with reduced errors,
S. F. Boys and F. Bernardi, “The calculation of small molecular inter- actions by the differences of separate total energies. some procedures with reduced errors,”Molecular Physics, vol. 19, no. 4, pp. 553–566, 1970
1970
-
[25]
A consistent and accu- rate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu,
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, “A consistent and accu- rate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu,”The Journal of Chemical Physics, vol. 132, no. 15, p. 154104, 2010
2010
-
[26]
Quantum information-assisted complete active space optimization (qicas),
L. Ding, S. Knecht, and C. Schilling, “Quantum information-assisted complete active space optimization (qicas),”The Journal of Physical Chemistry Letters, vol. 14, no. 49, pp. 11 022–11 029, 12 2023. [Online]. Available: https://doi.org/10.1021/acs.jpclett.3c02536
-
[27]
Qiskit addon: sample-based quantum diagonalization,
A. A. Saki, S. Barison, B. Fuller, J. R. Garrison, J. R. Glick, C. Johnson, A. Mezzacapo, J. Robledo-Moreno, M. Rossmannek, P. Schweigert, I. Sitdikov, and K. J. Sung, “Qiskit addon: sample-based quantum diagonalization,” https://github.com/Qiskit/qiskit-addon-sqd, 2024
2024
-
[28]
Sample-based quantum diagonalization (SQD): Qiskit addons documentation,
IBM Quantum, “Sample-based quantum diagonalization (SQD): Qiskit addons documentation,” https://quantum.cloud.ibm.com/docs/en/guides/ qiskit-addons-sqd, 2025, accessed: 2026-05-25
2025
-
[29]
K. Sugisaki, “Size-consistent implementation of hamiltonian simulation- based quantum-selected configuration interaction method for the supramolecular approach,”arXiv preprint arXiv:2510.23154, 2025
-
[30]
Hydrogen bond equilibria of phenol-pyridine in cyclohexane, carbon tetrachloride, and benzene solvents,
J. N. Spencer, J. C. Andrefsky, A. Grushow, J. Naghdi, L. M. Patti, and J. F. Trader, “Hydrogen bond equilibria of phenol-pyridine in cyclohexane, carbon tetrachloride, and benzene solvents,”The Journal of Physical Chemistry, vol. 91, no. 6, pp. 1673–1674, 1987
1987
-
[31]
Accurate quantum-centric simulations of supramolec- ular interactions,
D. Kaliakinet al., “Accurate quantum-centric simulations of supramolec- ular interactions,”arXiv preprint arXiv:2410.09209, 2024
-
[32]
How to use quantum computers for biomolecular free energies
J. G ¨unther, T. Weymuth, M. Bensberg, F. Witteveen, M. S. Teynor, F. E. Thomasen, V . Sora, W. Bro-Jørgensen, R. T. Husistein, M. Erakovic, M. Miller, L. Weisburn, M. Cho, M. Eckhoff, A. W. Harrow, A. Krogh, T. Van V oorhis, K. Lindorff-Larsen, G. Solomon, M. Reiher, and M. Christandl, “How to use quantum computers for biomolecular free energies,”arXiv p...
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[33]
K. S. V . Anuraget al., “Towards chemically accurate and scalable quantum simulations on IQM quantum hardware: A quantum-hpc hybrid approach,”arXiv preprint arXiv:2604.01983, 2026
-
[34]
Convergence of sample- based quantum diagonalization on a variable-length cuprate chain,
L. A. Wray, C.-J. Lin, V . Su, and H. Gharibyan, “Convergence of sample- based quantum diagonalization on a variable-length cuprate chain,” arXiv preprint arXiv:2512.04962, 2025
-
[35]
Advances in quantum computation in nisq era,
X.-D. Xie, X. Zhang, B. Koczor, and X. Yuan, “Advances in quantum computation in nisq era,”Entropy, vol. 27, no. 10, p. 1074, 2025
2025
-
[36]
A generally applicable atomic-charge dependent london dispersion correction,
E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth, and S. Grimme, “A generally applicable atomic-charge dependent london dispersion correction,”The Journal of Chemical Physics, vol. 150, no. 15, p. 154122, 2019
2019
-
[37]
Critical limitations in quantum-selected configuration interaction methods,
P. Reinholdt, K. M. Ziems, E. R. Kjellgren, S. Coriani, S. P. A. Sauer, and J. Kongsted, “Critical limitations in quantum-selected configuration interaction methods,”Journal of Chemical Theory and Computation, vol. 21, no. 14, pp. 6811–6822, 2025
2025
-
[38]
Noise- resilient quantum chemistry with half the qubits,
S. McFarthing, A. Pellow-Jarman, and F. Petruccione, “Noise- resilient quantum chemistry with half the qubits,”arXiv preprint arXiv:2602.01165, 2026
-
[39]
Hamil- tonian simulation-based quantum-selected configuration interaction for large-scale electronic structure calculations with a quantum computer,
K. Sugisaki, S. Kanno, T. Itoko, R. Sakuma, and N. Yamamoto, “Hamil- tonian simulation-based quantum-selected configuration interaction for large-scale electronic structure calculations with a quantum computer,” Physical Chemistry Chemical Physics, vol. 27, pp. 20 869–20 884, 2025
2025
-
[40]
A. K. Patra, K. S. V . Anurag, P. Sai Shankar, R. Bhat, V . Raghaven- dra, R. Maitra, and G. Jaiganesh, “Quantum simulation of ligand-like molecules through sample-based quantum diagonalization in density matrix embedding framework,”arXiv preprint arXiv:2511.22158, 2025
work page internal anchor Pith review Pith/arXiv arXiv 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.