SEDACS: A Scalable Framework for Complex Chemistry Simulations
Pith reviewed 2026-06-28 08:25 UTC · model grok-4.3
The pith
SEDACS enables stable long-time quantum molecular dynamics on systems of tens of thousands of atoms by decomposing electronic connectivity graphs into parallel overlapping subgraphs.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
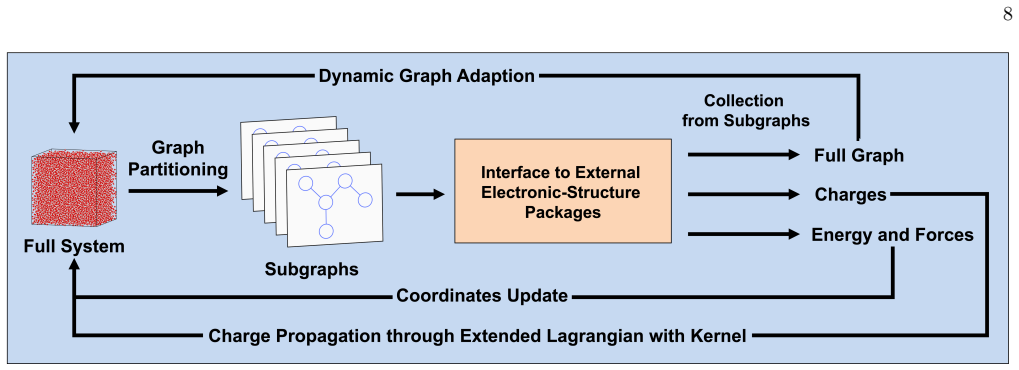
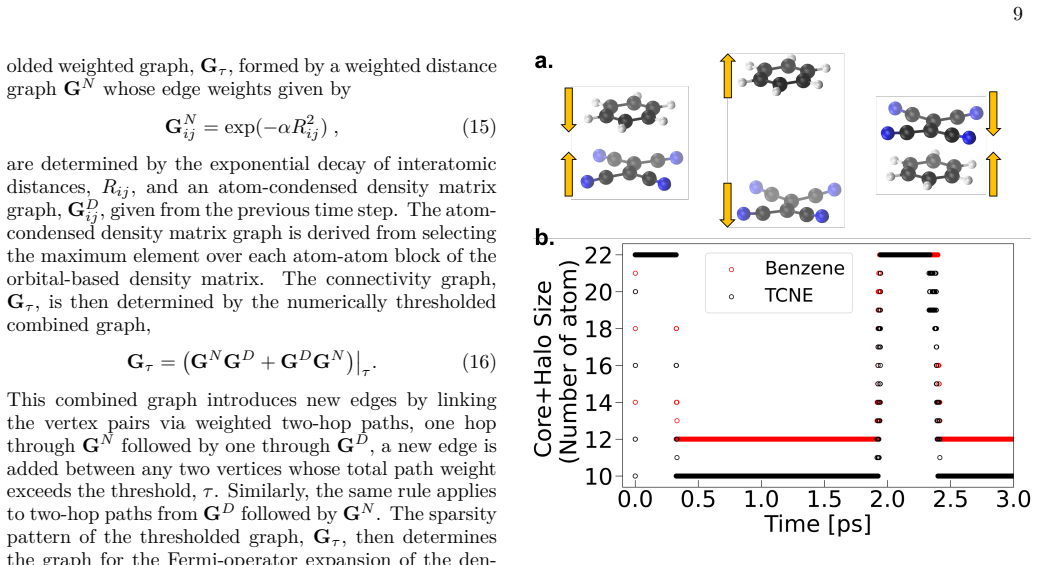
SEDACS integrates tunable graph construction, where edges encode non-local electronic overlap, with decomposition into overlapping subgraphs solved independently in parallel; when paired with shadow molecular dynamics it produces stable quantum molecular dynamics trajectories for systems containing tens of thousands of atoms, and does so by interfacing with external electronic-structure solvers through minimal code changes.
What carries the argument
The adaptive graph whose edges represent non-local electronic overlap, decomposed into smaller overlapping subgraphs that are solved independently and in parallel.
If this is right
- Stable QMD trajectories become feasible for chemically active systems with tens of thousands of atoms.
- Existing electronic-structure packages can be coupled to the framework with only minimal modifications.
- The approach supports a range of external solvers through its modular interface design.
- Long-time stability is maintained by the combination with extended-Lagrangian shadow molecular dynamics.
Where Pith is reading between the lines
- The same decomposition strategy might be tested on systems near the metallic boundary to map where the nearsightedness assumption begins to fail.
- Adaptive overlap tuning could be explored as a route to further error reduction in highly reactive local regions.
- Coupling the framework to multiple external codes simultaneously could enable mixed-accuracy simulations within a single trajectory.
Load-bearing premise
The nearsightedness of electronic connectivity in non-metallic systems ensures that independent solutions on overlapping subgraphs retain enough accuracy for chemically active systems.
What would settle it
A benchmark on a chemically active system of several thousand atoms in which total energy or atomic forces from the subgraph decomposition differ from a full calculation by more than chemical accuracy.
Figures







read the original abstract
Graph-based linear-scaling electronic-structure theory provides a scalable framework for parallel quantum-mechanical molecular dynamics (QMD) simulations by exploiting the nearsightedness of the non-local electronic connectivity in non-metallic systems. When combined with recent shadow molecular dynamics in an extended-Lagrangian formulation, it enables stable long-time simulations of large, chemically active systems. This article introduces the Scalable Ecosystem, Driver, and Analyzer for Complex Chemistry Simulations (SEDACS), which integrates all these advances within a modular, Python-based software package for large-scale QMD simulations driven by external electronic-structure codes. SEDACS provides a tunable, adaptive graph construction in which edges encode the non-local electronic overlap between atoms. This graph is then decomposed into a set of smaller, overlapping subgraphs, where the electronic structure of each of these subgraphs is solved for independently and in parallel using an external electronic-structure code. SEDACS can be coupled to a variety of external electronic-structure solvers with minimal modifications to their software, enabling rapid adoption of the graph-based QMD approach. In this way, SEDACS can greatly extend the capability of existing electronic-structure packages by enabling stable QMD simulations of systems that were previously computationally inaccessible. We demonstrate highly efficient and stable QMD simulations for chemically active systems with tens of thousands of atoms by interfacing SEDACS with an external Fortran-based electronic-structure code based on self-consistent-charge density functional tight-binding theory.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SEDACS, a modular Python-based framework integrating graph-based linear-scaling electronic-structure methods (exploiting nearsightedness of non-local electronic connectivity for decomposition into overlapping subgraphs) with shadow molecular dynamics. It enables coupling to external electronic-structure codes with minimal changes and demonstrates stable, efficient QMD simulations of chemically active systems with tens of thousands of atoms using SCC-DFTB.
Significance. If the accuracy of the subgraph decomposition holds, SEDACS would meaningfully extend existing electronic-structure packages to large-scale QMD simulations of complex systems previously inaccessible due to computational cost. The modular design and emphasis on external-code compatibility are practical strengths that could aid adoption.
major comments (2)
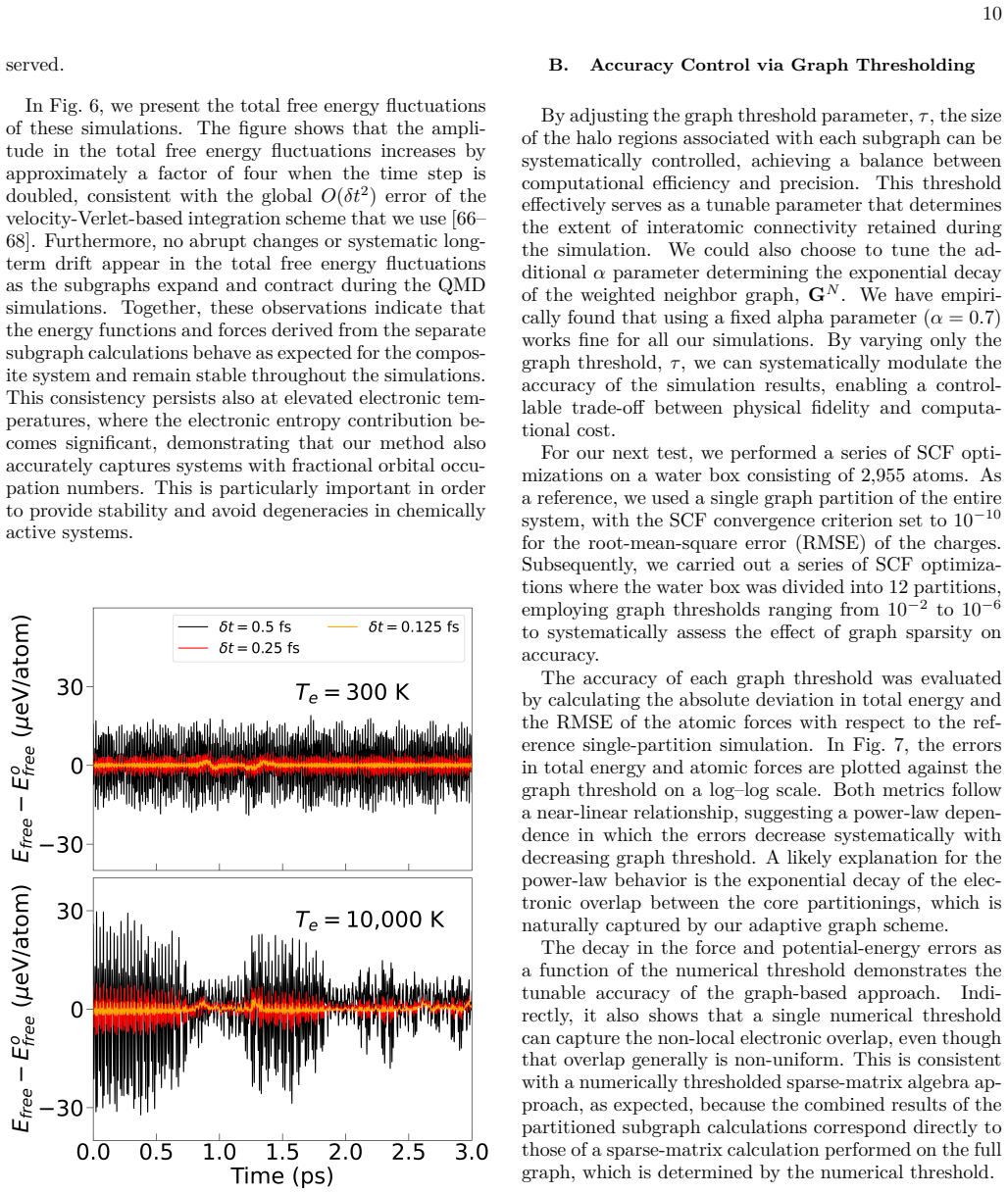
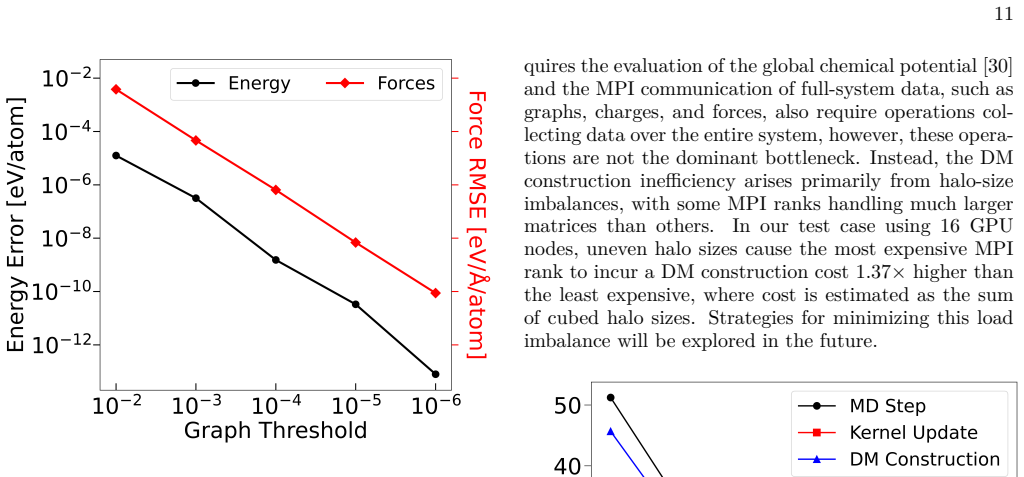
- [Abstract] Abstract and demonstration paragraph: the claim of stable QMD for chemically active systems with tens of thousands of atoms reports only stability and scaling but provides no quantitative error metrics (e.g., ΔE or force RMSE) versus full-system reference calculations on the same systems where both are feasible. This directly bears on whether the graph decomposition preserves sufficient accuracy.
- [Abstract] Paragraph on graph construction: the central assumption that nearsightedness of electronic connectivity allows independent subgraph solves to retain accuracy for chemically active non-metallic systems is invoked but not tested with direct comparisons to undecomposed calculations in the regime of the demonstration.
minor comments (1)
- The manuscript would benefit from explicit reporting of the overlap sizes, graph-construction parameters, and any convergence tests used in the tens-of-thousands-atom demonstration to support reproducibility.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We address each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Abstract] Abstract and demonstration paragraph: the claim of stable QMD for chemically active systems with tens of thousands of atoms reports only stability and scaling but provides no quantitative error metrics (e.g., ΔE or force RMSE) versus full-system reference calculations on the same systems where both are feasible. This directly bears on whether the graph decomposition preserves sufficient accuracy.
Authors: We agree that quantitative error metrics are important for validating accuracy. The current manuscript prioritizes demonstrating stability and scaling on systems (tens of thousands of atoms) where full-system references are infeasible. We have performed direct comparisons (energy and force errors) versus undecomposed calculations on smaller systems up to a few thousand atoms where both are feasible, and these show errors remain small. In the revised manuscript we will add these quantitative results (e.g., ΔE and force RMSE tables or plots) to the main text or a dedicated validation section, along with discussion of how they support the large-system claims. revision: yes
-
Referee: [Abstract] Paragraph on graph construction: the central assumption that nearsightedness of electronic connectivity allows independent subgraph solves to retain accuracy for chemically active non-metallic systems is invoked but not tested with direct comparisons to undecomposed calculations in the regime of the demonstration.
Authors: We acknowledge that explicit tests of the nearsightedness assumption via direct comparisons strengthen the central claim. The demonstration regime involves systems too large for full calculations, but we have conducted such tests on representative smaller and medium-sized chemically active non-metallic systems. We will incorporate these direct comparison results into the revised manuscript (new subsection or appendix) to explicitly show accuracy retention under the graph decomposition, thereby supporting the method's applicability to the larger demonstration cases. revision: yes
Circularity Check
No circularity: software integration framework with no self-referential derivations or fitted predictions.
full rationale
The paper describes SEDACS as a modular Python package that couples graph decomposition (based on the established nearsightedness principle) to external electronic-structure solvers for parallel QMD. No equations, parameters, or physical predictions are derived within the manuscript; the contribution is an integration layer and demonstration of scaling/stability on systems up to tens of thousands of atoms. The nearsightedness assumption is invoked as prior knowledge rather than derived or fitted here, and no self-citation chain is used to justify a uniqueness result or ansatz. The work is therefore self-contained as an engineering framework without any reduction of outputs to inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Car and M
R. Car and M. Parrinello, Phys. Rev. Lett.55, 2471 (1985)
1985
-
[2]
Allen and D
M. Allen and D. Tildesley,Computer Simulation of Liq- uids(Oxford Science, London, 1990)
1990
-
[3]
Carloni, U
P. Carloni, U. Rothlisberger, and M. Parrinello, Acc. Chem. Res.35, 455 (2002)
2002
-
[4]
J. S. Tse, Annu. Rev. Phys. Chem.53, 249 (2002)
2002
-
[5]
A. F. Voter and F. M. ad T. C. Germann, Annu. Rev. Mater. Res.32, 321 (2002)
2002
-
[6]
R. Iftimie, P. Minary, and M. E. Tuckerman, Proc. Natl. Acad. Sci. U.S.A.102, 6654 (2005), https://www.pnas.org/doi/pdf/10.1073/pnas.0500193102
-
[7]
Marx and J
D. Marx and J. Hutter,Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods(Cambridge Uni- versity Press, 2009)
2009
-
[8]
M. E. Tuckerman,Statistical Mechanics: Theory and Molecular Simulation(Oxford University Press, New York, 2010)
2010
-
[9]
Karplus, Angew
M. Karplus, Angew. Chem. Int. Ed.53, 9992 (2014)
2014
-
[10]
W. Jia, H. Wang, M. Chen, D. Lu, L. Lin, R. Car, E. Weinan, and L. Zhang, inSC20: International Con- ference for High Performance Computing, Networking, Storage and Analysis(2020) pp. 1–14
2020
-
[11]
Fedik, R
N. Fedik, R. Zubatyuk, M. Kulichenko, N. Lubbers, J. S. Smith, B. Nebgen, R. Messerly, Y. W. Li, A. I. Boldyrev, K. Barros, O. Isayev, and S. Tretiak, Nat. Rev. Chem. 6, 653 (2022)
2022
-
[12]
Zhang, V
Y.-W. Zhang, V. Sorkin, Z. H. Aitken, A. Politano, J. Behler, A. P Thompson, T. W. Ko, S. P. Ong, O. Cha- lykh, D. Korogod, E. Podryabinkin, A. Shapeev, J. Li, Y. Mishin, Z. Pei, X. Liu, J. Kim, Y. Park, S. Hwang, S. Han, K. Sheriff, Y. Cao, and R. Freitas, Modelling Simul. Mater. Sci. Eng.33, 023301 (2025). 14
2025
-
[13]
M. Kulichenko, B. Nebgen, N. Lubbers, J. S. Smith, K. Barros, A. E. A. Allen, A. Habib, E. Shinkle, N. Fedik, Y. W. Li, R. A. Messerly, and S. Tre- tiak, Chem. Rev.124, 13681 (2024), pMID: 39572011, https://doi.org/10.1021/acs.chemrev.4c00572
-
[14]
Ouyang, X
Y. Ouyang, X. Chen, Y. Liu, X. Chen, H. Shang, Z. Chen, R. Lin, X. Gao, L. Wang, F. Li, J. Shan, H. Song, H. Cui, X. Feng, and J. Xue, inProceedings of the Inter- national Conference for High Performance Computing, Networking, Storage and Analysis, SC ’25 (Association for Computing Machinery, New York, NY, USA, 2025) p. 1631–1645
2025
-
[15]
Kalita, H
B. Kalita, H. Gokcan, and O. Isayev, Nat. Comput. Sci. 5, 1120 (2025)
2025
-
[16]
Hohenberg and W
P. Hohenberg and W. Kohn, Phys. Rev.136, B:864 (1964)
1964
-
[17]
Kohn and L
W. Kohn and L. J. Sham, Phys. Rev.140, 1133 (1965)
1965
-
[18]
R. G. Parr and W. Yang,Density-functional theory of atoms and molecules(Oxford University Press, Oxford, 1989)
1989
-
[19]
Dreizler and K
R. Dreizler and K. Gross,Density-functional theory (Springer Verlag, Berlin Heidelberg, 1990)
1990
-
[20]
Goedecker, Rev
S. Goedecker, Rev. Mod. Phys.71, 1085 (1999)
1999
-
[21]
D. R. Bowler and T. Miyazaki, J. Phys.: Condens. Matter 22, 074207 (2010)
2010
-
[22]
D. R. Bowler and T. Miyazaki, Rep. Prog. Phys.75, 036503 (2012)
2012
-
[23]
S. Mohr, L. E. Ratcliff, L. Genovese, D. Caliste, P. Boulanger, S. Goedecker, and T. Deutsch, Phys. Chem. Chem. Phys.17, 31360 (2015)
2015
-
[24]
Fattebert, D
J.-L. Fattebert, D. Osei-Kuffuor, E. W. Draeger, T. Og- itsu, and W. D. Krauss, inSC ’16: Proceedings of the International Conference for High Performance Comput- ing, Networking, Storage and Analysis(2016) pp. 12–22
2016
-
[25]
Elstner, D
M. Elstner, D. Poresag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai, and G. Seifert, Phys. Rev. B58, 7260 (1998)
1998
-
[26]
M. W. Finnis, A. T. Paxton, M. Methfessel, and M. van Schilfgarde, Phys. Rev. Lett.81, 5149 (1998)
1998
-
[27]
Frauenheim, G
T. Frauenheim, G. Seifert, M. Elstner, Z. Hajnal, G. Jungnickel, D. Poresag, S. Suhai, and R. Scholz, Phys. Stat. sol.217, 41 (2000)
2000
-
[28]
Hourahine, B
B. Hourahine, B. Aradi, V. Blum, F. Bonaf´ e, A. Buc- cheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Du- mitric˘ a, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. K¨ ohler, T. Kowalczyk, T. Kubaˇ r, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J...
2020
-
[29]
A. M. N. Niklasson, S. M. Mnizsewski, C. F. A. Negre, M. J. Cawkwell, P. J. Swart, J. Mohd-Yusof, T. C. Ger- mann, M. E. Wall, N. Bock, E. H. Rubensson, and H. N. Djidjev, J. Chem. Phys.144, 234101 (2016)
2016
-
[30]
C. F. A. Negre, M. E. Wall, and A. M. N. Niklasson, J. Chem. Phys.158, 074108 (2023)
2023
-
[31]
Graph parti- tioning methods for fast parallel quantum molecular dy- namics,
H. N. Djidjev, G. Hahn, S. M. Mniszewski, C. F. Negre, A. M. Niklasson, and V. B. Sardeshmukh, “Graph parti- tioning methods for fast parallel quantum molecular dy- namics,” in2016 Proceedings of the Seventh SIAM Work- shop on Combinatorial Scientific Computing, pp. 42–51, http781611974690.ch5
-
[32]
M. Lass, S. Mohr, H. Wiebeler, T. K¨ uhne, and C. Plessl, inProceedings of the Platform for Advanced Scientific Computing (PASC) Conference(ACM, 2018)
2018
-
[33]
H. N. Djidjev, G. Hahn, S. M. Mniszewski, C. F. A. Negre, and A. M. N. Niklasson, Algorithms12(2019), 10.3390/a12090187
-
[34]
M. Lass, R. Schade, T. K¨ uhne, and C. Plessl, inPro- ceedings of the International Conference for High Per- formance Computing, Networking, Storage and Analysis (SC)(IEEE Computer Society, 2020) p. 1127
2020
-
[35]
Kulichenko, R
M. Kulichenko, R. M. Stanton, C.-H. Li, N. Lubbers, S. Tretiak, M. E. Wall, C. F. Negre, J. Finkelstein, and A. M. Niklasson, Journal of Chemical Theory and Com- putation21, 10780 (2025)
2025
-
[36]
W. T. Yang, Phys. Rev. Lett.66, 1438 (1991)
1991
-
[37]
W. T. Yang and T. S. Lee, J. Chem. Phys.103, 5674 (1995)
1995
-
[38]
Kitaura, E
K. Kitaura, E. Ikeo, T. Asada, T. Nakano, and M. Ue- bayasi, Chemical Physics Letters313, 701 (1999)
1999
-
[39]
Ozaki, Phys
T. Ozaki, Phys. Rev. B74, 245101 (2006)
2006
-
[40]
V. Q. Vuong, Y. Nishimoto, D. G. Fedorov, B. G. Sumpter, T. A. Niehaus, and S. Irle, Journal of Chem- ical Theory and Computation15, 3008 (2019), pMID: 30998360, https://doi.org/10.1021/acs.jctc.9b00108
-
[41]
The fmo-dftb method,
Y. Nishimoto and S. Irle, “The fmo-dftb method,” inRecent Advances of the Fragment Molecular Orbital Method: Enhanced Performance and Applicability, edited by Y. Mochizuki, S. Tanaka, and K. Fukuzawa (Springer Singapore, Singapore, 2021) pp. 459–485
2021
-
[42]
A. H. R. Palser and D. E. Manolopoulos, Phys. Rev. B 58, 12704 (1998)
1998
-
[43]
A. M. N. Niklasson, Phys. Rev. B66, 155115 (2002)
2002
-
[44]
E. H. Rubensson and A. M. N. Niklasson, SIAM J. Sci. Comput.36, 148 (2014)
2014
-
[45]
L. A. Truflandier, R. M. Dianzinga, and D. R. Bowler, J. Chem. Phys.144(2016), 10.1063/1.4943213
-
[46]
Borstnik, J
U. Borstnik, J. VandeVondele, V. Weber, and J. Hutter, Parallel Comput.40, 47 (2014)
2014
-
[47]
Weber, T
V. Weber, T. Latino, A. Pozdeev, I. Feduova, and A. Cu- rioni, J. Chem. Theory Comput.11, 3145 (2015)
2015
-
[48]
and Neese, Frank , year = 2015, month = jul, journal =
P. Pinski, C. Riplinger, E. F. Valeev, and F. Neese, The Journal of Chemical Physics143, 034108 (2015), https://doi.org/10.1063/1.4926879
-
[49]
A. Kruchinina, E. Rudberg, and E. H. Rubens- son, Journal of Chemical Theory and Com- putation12, 5788 (2016), pMID: 27783507, https://doi.org/10.1021/acs.jctc.6b00626
-
[50]
J. Finkelstein, J. S. Smith, S. M. Mniszewski, K. Bar- ros, C. F. A. Negre, E. H. Rubensson, and A. M. N. Niklasson, Journal of Chemical Theory and Computation17, 2256 (2021), pMID: 33797253, https://doi.org/10.1021/acs.jctc.1c00057
-
[51]
A. M. N. Niklasson, Phys. Rev. Lett.100, 123004 (2008)
2008
-
[52]
Hirakawa, T
T. Hirakawa, T. suzuki, D. R. Bowler, and T. Myazaki, J. Phys.: Condens. Matter29, 405901 (2017)
2017
-
[53]
A. M. N. Niklasson, Eur. Phys. J. B94, 164 (2021)
2021
-
[54]
A. M. N. Niklasson and C. F. A. Negre, The Journal of Chemical Physics158, 154105 (2023)
2023
-
[55]
M. J. Cawkwell and et al., “LATTE,” (2010), Los Alamos National Laboratory (LA- CC-10004), http://www.github.com/lanl/latte
2010
-
[56]
N. Bock, M. J. Cawkwell, J. D. Coe, A. Krishnapriyan, M. P. Kroonblawd, A. Lang, C. Liu, E. M. Saez, S. M. Mniszewski, C. F. A. Negre, A. M. N. Niklas- 15 son, E. Sanville, M. A. Wood, and P. Yang, “LATTE,” (2008)
2008
-
[57]
Perriot, C
R. Perriot, C. F. A. Negre, S. D. McGrane, and M. J. Cawkwell, AIP Conf. Proc.1979, 050014 (2018)
1979
-
[58]
J. Finkelstein, E. H. Rubensson, S. M. Mniszewski, C. F. A. Negre, and A. M. N. Niklas- son, Journal of Chemical Theory and Com- putation18, 4255 (2022), pMID: 35670603, https://doi.org/10.1021/acs.jctc.2c00274
-
[59]
Karypis, , and V
G. Karypis, , and V. Kumar, SIAM J. Sci. Comput.20, 359 (1999)
1999
-
[60]
D. K. Remler and P. A. Madden, Mol. Phys.70, 921 (1990)
1990
-
[61]
Pulay and G
P. Pulay and G. Fogarasi, Chem. Phys. Lett.386, 272 (2004)
2004
-
[62]
A. M. N. Niklasson, C. J. Tymczak, and M. Challa- combe, Phys. Rev. Lett.97, 123001 (2006)
2006
-
[63]
A. M. N. Niklasson, J. Chem. Phys.147, 054103 (2017)
2017
-
[64]
A. M. N. Niklasson, J. Chem. Phys.152, 104103 (2020)
2020
-
[65]
A. M. N. Niklasson, J. Chem. Theory Comput.16, 3628 (2020)
2020
-
[66]
Steneteg, I
P. Steneteg, I. A. Abrikosov, V. Weber, and A. M. N. Niklasson, Phys. Rev. B82, 075110 (2010)
2010
-
[67]
A. M. N. Niklasson, P. Steneteg, A. Odell, N. Bock, M. Challacombe, C. J. Tymczak, E. Holmstrom, G. Zheng, and V. Weber, J. Chem. Phys.130, 214109 (2009)
2009
-
[68]
Zheng, A
G. Zheng, A. M. N. Niklasson, and M. Karplus, J. Chem. Phys.135, 044122 (2011)
2011
-
[69]
Pulay, Chem
P. Pulay, Chem. Phys. Let.73, 393 (1980)
1980
-
[70]
Pulay, J
P. Pulay, J. Comput. Chem.3, 556 (1982)
1982
-
[71]
Habib, J
A. Habib, J. Finkelstein, and A. M. Niklasson, Journal of Chemical Theory and Computation20, 7102 (2024)
2024
-
[72]
Gpu-accelerated charge- equilibration for shadow molecular dynamics in python,
M. C. Kaymak, N. Lubbers, C. F. A. Negre, M. Wall, and A. M. N. Niklasson, “Gpu-accelerated charge- equilibration for shadow molecular dynamics in python,” (2025), arXiv:2503.21176 [physics.comp-ph]
arXiv 2025
-
[73]
Woi´ nska, S
M. Woi´ nska, S. Grabowsky, P. M. Dominiak, K. Wo´ zniak, and D. Jayatilaka, Sci. Adv.2, e1600192 (2016)
2016
-
[74]
Strachan, A
A. Strachan, A. C. T. van Duin, D. Chakraborty, S. Das- gupta, and W. A. Goddard, Phys. Rev. Lett.91, 098301 (2003)
2003
-
[75]
J. Wu, J. Wu, J. Li, Y. Shang, and L. Chen, ACS Omega8, 18851 (2023), https://doi.org/10.1021/acsomega.3c01160
-
[76]
A. M. N. Niklasson, M. J. Cawkwell, E. H. Rubensson, and E. Rudberg, Phys. Rev. E92, 063301 (2015)
2015
-
[77]
Nishimoto, J
Y. Nishimoto, J. Chem. Phys.146, 084101 (2017)
2017
-
[78]
Porezag, T
D. Porezag, T. Frauenheim, T. K¨ ohler, G. Seifert, and R. Kaschner, Phys. Rev. B51, 12947 (1995)
1995
-
[79]
Seifert, D
G. Seifert, D. Porezag, and T. Frauenheim, International Journal of Quantum Chemistry58, 185 (1996)
1996
-
[80]
M. Gaus, Q. Cui, and M. Elstner, J, Chem. Theory Comput.7, 931 (2011)
2011
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.