Spatial Transcriptomics-Guided Alignment Enhances Molecular Profiling in Pathology Foundation Model
Pith reviewed 2026-06-28 23:24 UTC · model grok-4.3
The pith
STAMP endows pathology foundation models with molecular awareness by aligning them to spatial transcriptomics via pathway aggregation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
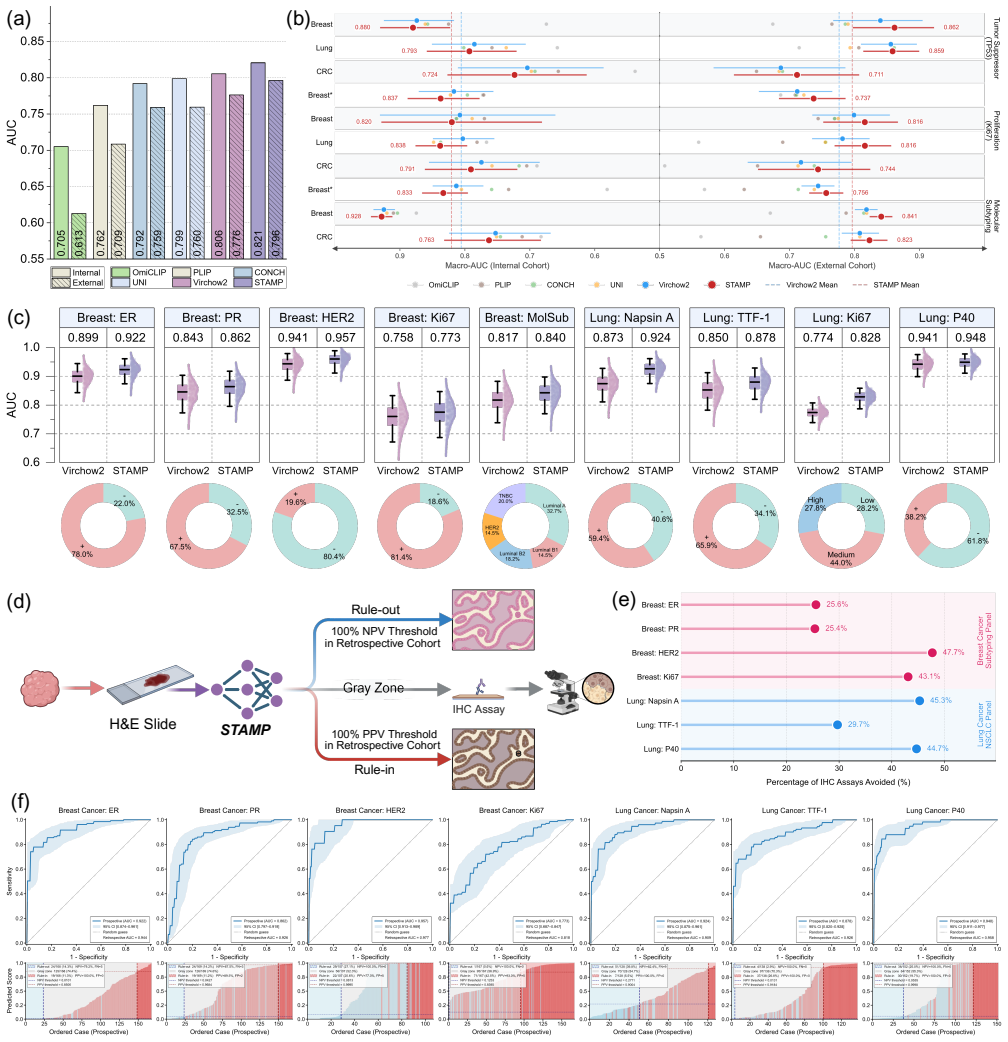
STAMP integrates pathway-aggregated spatial transcriptomics into PFMs through parameter-efficient fine-tuning, enriching the representation space and unlocking the capacity to resolve sub-visual molecular signatures from H&E WSIs.
What carries the argument
The pathway-informed alignment strategy that aggregates raw transcriptomic counts into biologically functional pathways before fine-tuning the PFMs.
If this is right
- PFMs can perform molecular profiling from routine H&E WSIs without direct sequencing.
- The models resolve sub-visual molecular signatures tied to histology.
- Clinical utility is shown through multi-tier evaluation on diverse anatomical sites and sequencing platforms.
- HumanST-1k supplies 1.8 million aligned image-transcriptomic pairs as a reusable training resource.
Where Pith is reading between the lines
- The same alignment method could be applied to other tissue imaging types to extract molecular signals.
- Improved molecular awareness might boost accuracy on downstream tasks such as treatment response prediction.
- Wider adoption could lower the frequency of repeat biopsies needed for molecular testing.
Load-bearing premise
Aggregating transcriptomic counts into pathways reduces technical noise enough to preserve the exact spatial link between morphological features and genomic alterations.
What would settle it
STAMP-tuned models show no gain over baseline PFMs when predicting held-out gene expression or pathway activity from H&E images across multiple organs.
Figures

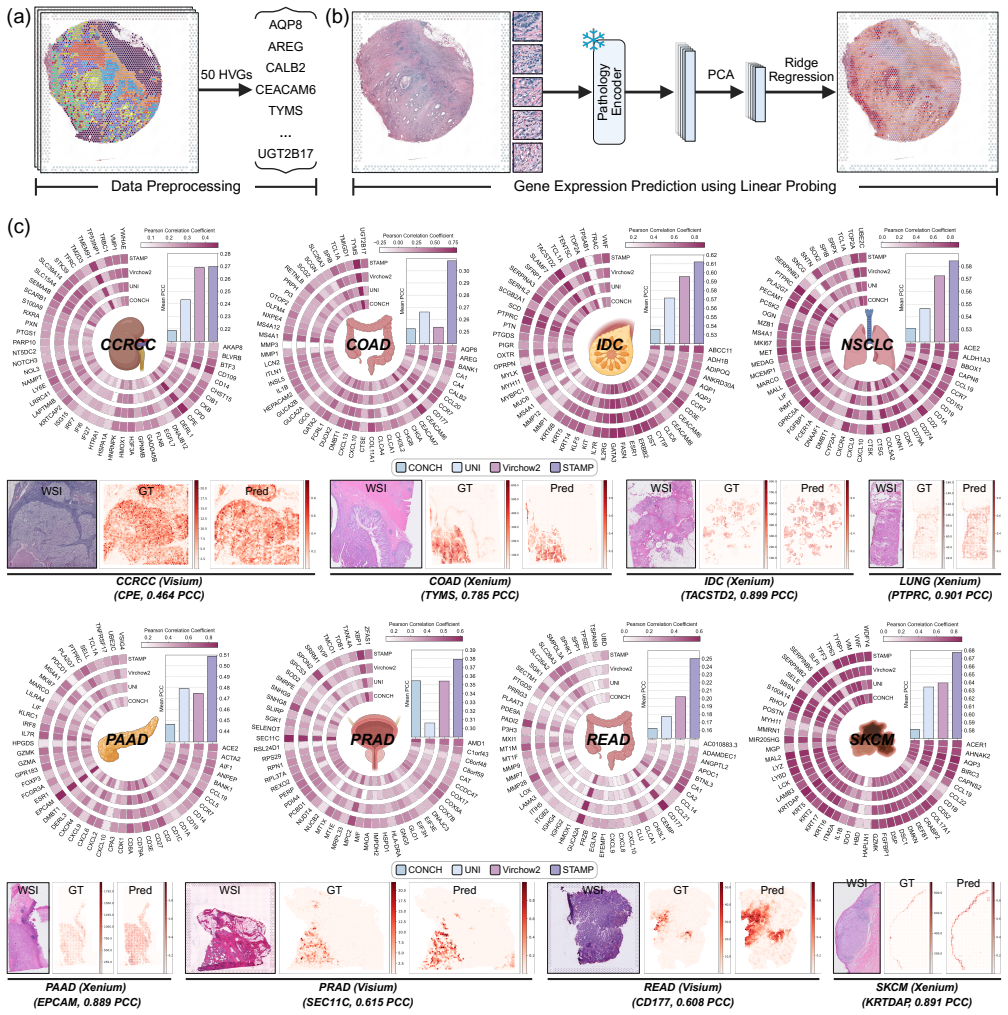
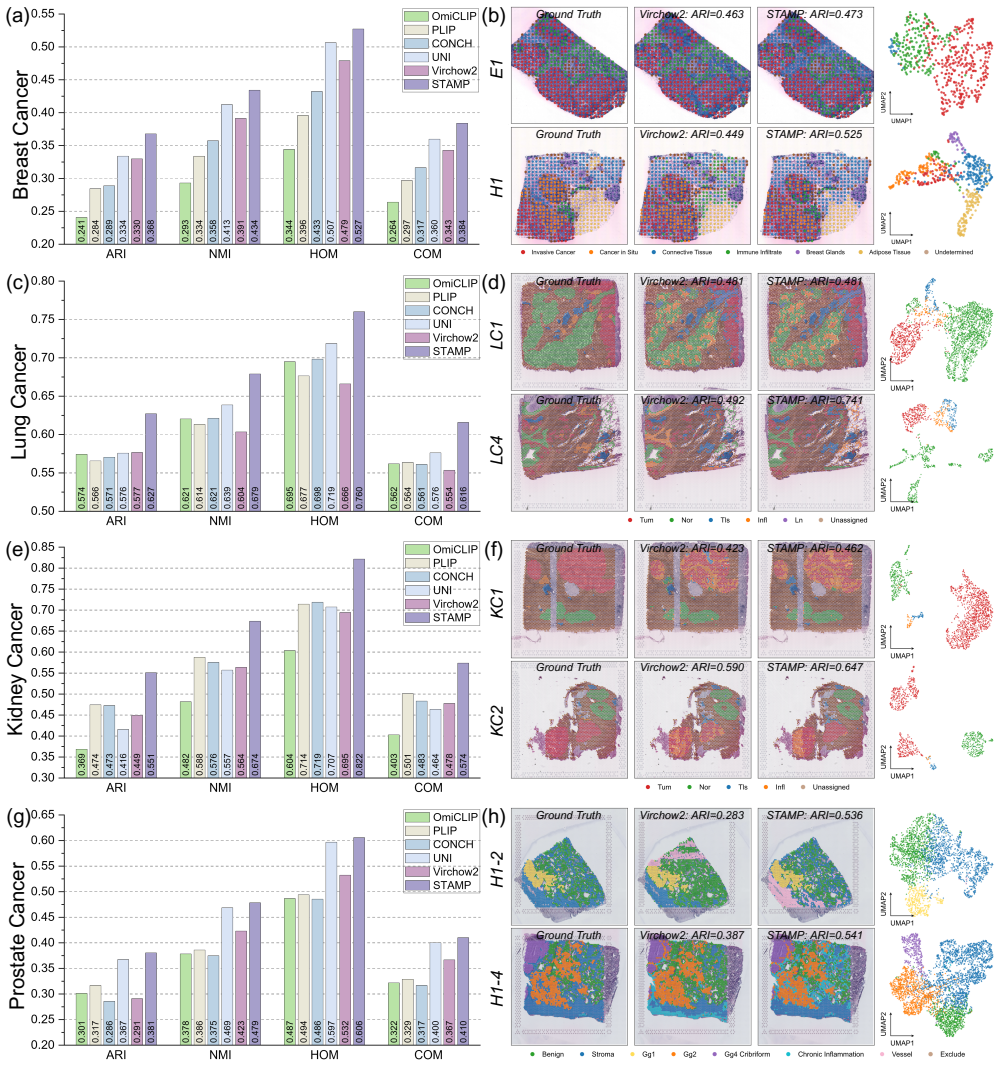
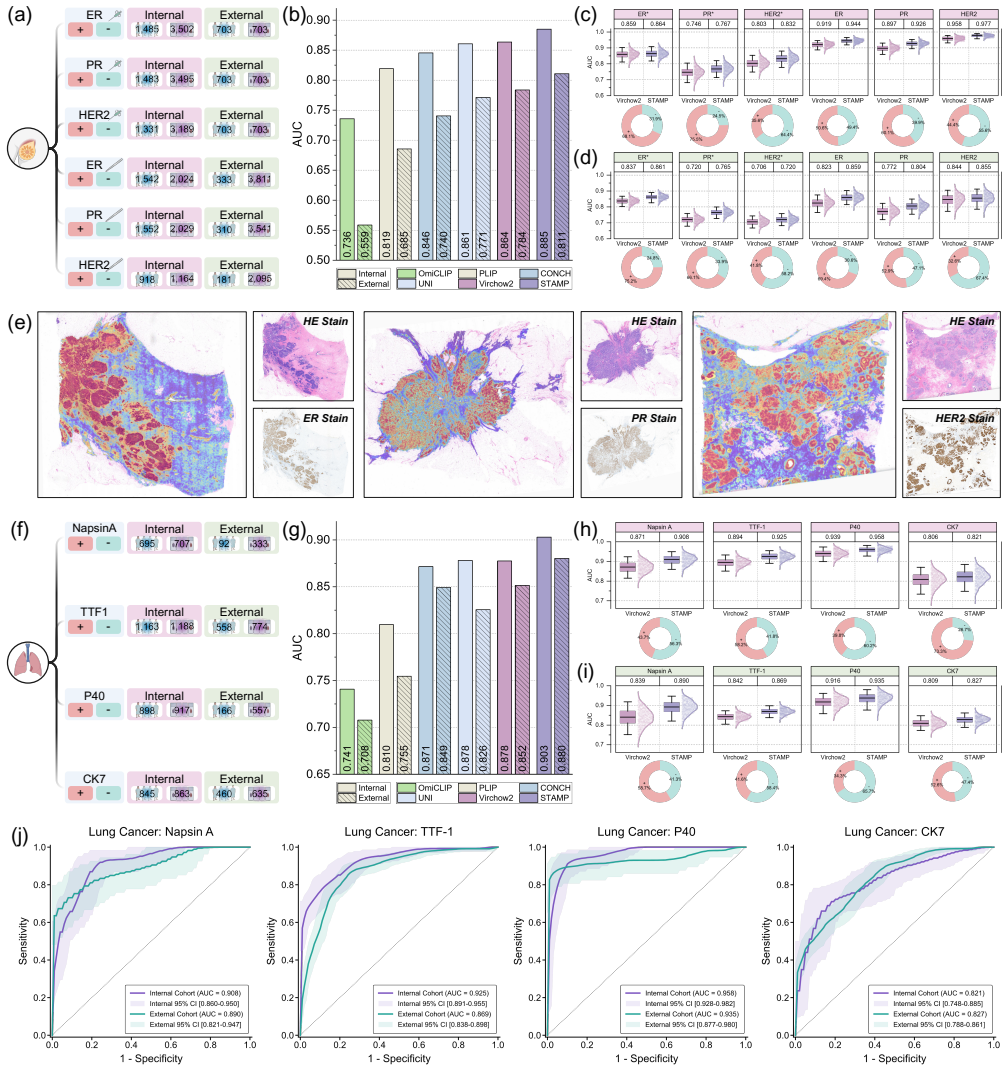
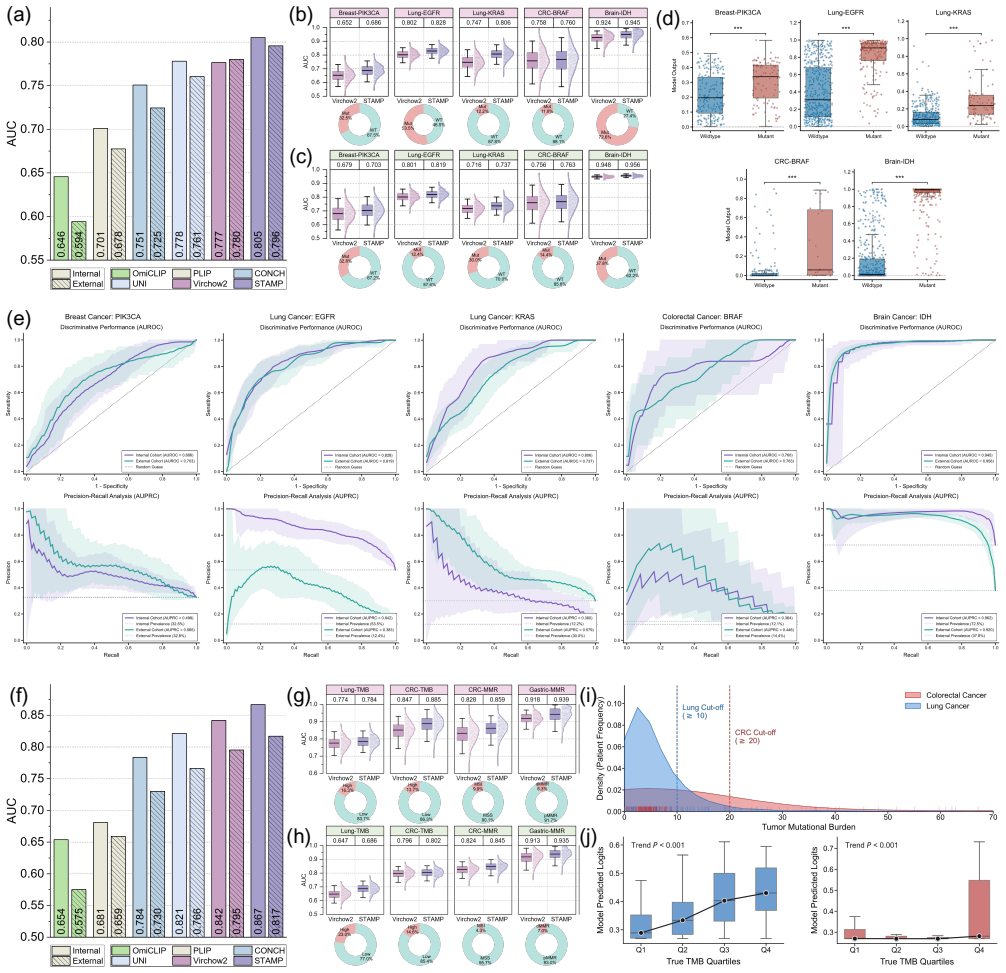
read the original abstract
Comprehensive molecular profiling is essential for modern precision oncology but remains hindered by prohibitive costs, specimen exhaustion, and protracted turnaround times. While pathology foundation models (PFMs) have demonstrated potential for inferring molecular phenotypes from routine hematoxylin and eosin (H&E) whole-slide images (WSIs), current architectures primarily rely on vision-centric self-supervised learning or vision-language alignment, lacking the spatially resolved molecular supervision required to connect subtle morphological features with underlying genomic alterations. Spatial transcriptomics (ST) emerges as a transformative technology that enables transcriptomic quantification within intact tissue sections, thereby preserving the precise spatial link between histology and molecular profiles. In this study, we present a Spatial Transcriptomics-guided Alignment framework for Molecular Profiling (STAMP), which endows PFMs with intrinsic molecular awareness. To support this paradigm, we curated HumanST-1k, a human ST dataset spanning diverse anatomical organs and sequencing platforms. This atlas yields 1.8 million pairs of H&E patches and corresponding transcriptomic profiles, providing a corpus that links histological structures with their molecular states. To mitigate the technical noise inherent to raw transcriptomics, STAMP applies a pathway-informed alignment strategy that aggregates transcriptomic data into biologically functional pathways, which are subsequently integrated into PFMs via parameter-efficient fine-tuning. This alignment enriches the representation space of PFMs and unlocks their capacity to resolve sub-visual molecular signatures. The clinical utility of these augmented representations was validated through a multi-tier evaluation framework.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces the STAMP framework, which endows pathology foundation models (PFMs) with intrinsic molecular awareness by curating the HumanST-1k dataset (1.8 million H&E patch–transcriptomic profile pairs across organs and platforms) and applying a pathway-informed alignment strategy. Raw spatial transcriptomics counts are aggregated into biologically functional pathways to mitigate technical noise, then integrated into PFMs via parameter-efficient fine-tuning; the resulting representations are claimed to resolve sub-visual molecular signatures from routine H&E WSIs, with clinical utility assessed via a multi-tier evaluation framework.
Significance. If the empirical results hold, the work would be significant for precision oncology: it directly addresses the lack of spatially resolved molecular supervision in current vision-centric or vision-language PFMs by leveraging ST data to link subtle morphology with genomic alterations. The scale of the HumanST-1k atlas and the practical use of parameter-efficient fine-tuning are clear strengths that could enable broader adoption and serve as a resource for the community.
major comments (1)
- [Abstract] Abstract: the central claim that pathway-informed aggregation 'mitigates the technical noise inherent to raw transcriptomics' while 'preserving the precise spatial link' between histology and molecular profiles is load-bearing for the entire alignment strategy. Aggregation necessarily collapses gene-level spatial variation; without any referenced quantitative check (e.g., patch-level mutual information between aggregated pathways and raw counts, or ablation against non-aggregated ST baselines), it remains unclear whether the procedure retains the sub-visual signatures the model is intended to resolve.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback on our manuscript. We address the single major comment below and will incorporate revisions to strengthen the justification for the pathway-informed aggregation strategy.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that pathway-informed aggregation 'mitigates the technical noise inherent to raw transcriptomics' while 'preserving the precise spatial link' between histology and molecular profiles is load-bearing for the entire alignment strategy. Aggregation necessarily collapses gene-level spatial variation; without any referenced quantitative check (e.g., patch-level mutual information between aggregated pathways and raw counts, or ablation against non-aggregated ST baselines), it remains unclear whether the procedure retains the sub-visual signatures the model is intended to resolve.
Authors: We agree that the pathway aggregation step is central to the STAMP framework and that explicit quantitative validation would strengthen the manuscript. The aggregation into Hallmark and Reactome pathways is motivated by established biological priors that reduce technical noise (e.g., dropout, batch effects) while retaining functional signals, but we acknowledge the absence of a direct patch-level comparison in the current version. In the revised manuscript we will add: (1) a supplementary analysis computing patch-level Pearson correlation and mutual information between raw gene counts and pathway aggregates across the HumanST-1k atlas; (2) an ablation study training the alignment module on raw counts versus pathway aggregates and reporting downstream performance on molecular phenotype prediction tasks. These additions will quantify whether the procedure preserves spatially relevant molecular variation. revision: yes
Circularity Check
No significant circularity; empirical pipeline is self-contained
full rationale
The paper describes a data curation step (HumanST-1k atlas yielding 1.8M H&E-transcript pairs), followed by pathway-informed aggregation of raw counts and parameter-efficient fine-tuning of existing PFMs. No equations, uniqueness theorems, fitted parameters renamed as predictions, or self-citation chains are present in the provided text. The central claim rests on empirical validation of the resulting representations rather than any derivation that reduces to its own inputs by construction. This is the expected non-finding for a methods-oriented empirical study.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
& Rubin, M
Akhoundova, D. & Rubin, M. A. Clinical application of advanced multi-omics tumor profiling: Shaping precision oncology of the future.Cancer cell40, 920–938 (2022)
2022
-
[2]
Cancers17, 3500 (2025)
Brlek, P.et al.Advances in precision oncology: From molecular profiling to regulatory-approved targeted therapies. Cancers17, 3500 (2025)
2025
-
[3]
A., Rimm, D
Bera, K., Schalper, K. A., Rimm, D. L., Velcheti, V . & Madabhushi, A. Artificial intelligence in digital pathology—new tools for diagnosis and precision oncology.Nat. reviews Clin. oncology16, 703–715 (2019)
2019
-
[4]
Niazi, M. K. K., Parwani, A. V . & Gurcan, M. N. Digital pathology and artificial intelligence.The lancet oncology20, e253–e261 (2019)
2019
-
[5]
N.et al.Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer
Kather, J. N.et al.Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. medicine25, 1054–1056 (2019). 7.Kather, J. N.et al.Pan-cancer image-based detection of clinically actionable genetic alterations.Nat. cancer1, 789–799 (2020)
2019
-
[6]
communications11, 3877 (2020)
Schmauch, B.et al.A deep learning model to predict rna-seq expression of tumours from whole slide images.Nat. communications11, 3877 (2020)
2020
-
[7]
Saltz, J.et al.Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images.Cell reports23, 181–193 (2018)
2018
-
[8]
Barkley, D.et al.Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat. genetics54, 1192–1201 (2022)
2022
-
[9]
Jerby-Arnon, L.et al.A cancer cell program promotes t cell exclusion and resistance to checkpoint blockade.Cell175, 984–997 (2018)
2018
-
[10]
J.et al.Towards a general-purpose foundation model for computational pathology.Nat
Chen, R. J.et al.Towards a general-purpose foundation model for computational pathology.Nat. medicine30, 850–862 (2024). 13.Xu, H.et al.A whole-slide foundation model for digital pathology from real-world data.Nature630, 181–188 (2024)
2024
-
[11]
medicine30, 2924–2935 (2024)
V orontsov, E.et al.A foundation model for clinical-grade computational pathology and rare cancers detection.Nat. medicine30, 2924–2935 (2024)
2024
-
[12]
Wang, X.et al.A pathology foundation model for cancer diagnosis and prognosis prediction.Nature634, 970–978 (2024)
2024
-
[13]
Ma, J.et al.A generalizable pathology foundation model using a unified knowledge distillation pretraining framework. Nat. Biomed. Eng.1–20 (2025). 17.Oquab, M.et al.Dinov2: Learning robust visual features without supervision.arXiv preprint arXiv:2304.07193(2023)
work page internal anchor Pith review Pith/arXiv arXiv 2025
-
[14]
InInternational conference on machine learning, 8748–8763 (PmLR, 2021)
Radford, A.et al.Learning transferable visual models from natural language supervision. InInternational conference on machine learning, 8748–8763 (PmLR, 2021)
2021
-
[15]
CoCa: Contrastive Captioners are Image-Text Foundation Models
Li, J., Li, D., Xiong, C. & Hoi, S. Blip: Bootstrapping language-image pre-training for unified vision-language understand- ing and generation. InInternational conference on machine learning, 12888–12900 (PMLR, 2022). 20.Yu, J.et al.Coca: Contrastive captioners are image-text foundation models.arXiv preprint arXiv:2205.01917(2022). 21.Lu, M. Y .et al.A vi...
work page internal anchor Pith review Pith/arXiv arXiv 2022
-
[16]
Huang, Z., Bianchi, F., Yuksekgonul, M., Montine, T. J. & Zou, J. A visual–language foundation model for pathology image analysis using medical twitter.Nat. medicine29, 2307–2316 (2023)
2023
-
[17]
neural information processing systems 36, 37995–38017 (2023)
Ikezogwo, W.et al.Quilt-1m: One million image-text pairs for histopathology.Adv. neural information processing systems 36, 37995–38017 (2023). 24.Xu, Y .et al.A multimodal knowledge-enhanced whole-slide pathology foundation model.Nat. Commun.(2025). 25.Marx, V . Method of the year: spatially resolved transcriptomics.Nat. methods18, 9–14 (2021)
2023
-
[18]
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics.Nature596, 211–220 (2021)
2021
-
[19]
national academy sciences102, 15545–15550 (2005)
Subramanian, A.et al.Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.Proc. national academy sciences102, 15545–15550 (2005). 28.Hu, E. J.et al.Lora: Low-rank adaptation of large language models.Iclr1, 3 (2022)
2005
-
[20]
Parameter-Efficient Fine-Tuning for Large Models: A Comprehensive Survey
Han, Z., Gao, C., Liu, J., Zhang, J. & Zhang, S. Q. Parameter-efficient fine-tuning for large models: A comprehensive survey.arXiv preprint arXiv:2403.14608(2024)
work page internal anchor Pith review Pith/arXiv arXiv 2024
- [21]
-
[22]
Ma, J.et al.Pathbench: A comprehensive comparison benchmark for pathology foundation models towards precision oncology.arXiv preprint arXiv:2505.20202(2025). 32.Liberzon, A.et al.The molecular signatures database hallmark gene set collection.Cell systems1, 417–425 (2015)
-
[23]
Methods22, 1568–1582 (2025)
Chen, W.et al.A visual–omics foundation model to bridge histopathology with spatial transcriptomics.Nat. Methods22, 1568–1582 (2025)
2025
-
[24]
Junttila, M. R. & De Sauvage, F. J. Influence of tumour micro-environment heterogeneity on therapeutic response.Nature 501, 346–354 (2013)
2013
-
[25]
medicine24, 541–550 (2018)
Binnewies, M.et al.Understanding the tumor immune microenvironment (time) for effective therapy.Nat. medicine24, 541–550 (2018)
2018
-
[26]
Roma-Rodrigues, C., Mendes, R., Baptista, P. V . & Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. Int. journal molecular sciences20, 840 (2019)
2019
-
[27]
Neural Inf
Jaume, G.et al.Hest-1k: A dataset for spatial transcriptomics and histology image analysis.Adv. Neural Inf. Process. Syst. 37, 53798–53833 (2024)
2024
-
[28]
Keren, L.et al.A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging.Cell174, 1373–1387 (2018)
2018
-
[29]
Andersson, A.et al.Spatial deconvolution of her2-positive breast tumors reveals novel intercellular relationships.bioRxiv 2020–07 (2020)
2020
-
[30]
Dawo, S., Nonchev, K. & Silina, K. 10x Visium Spatial Transcriptomics Dataset: Kidney (3) and Lung (5) Cancer with Tertiary Lymphoid Structures, DOI: 10.5281/zenodo.14620362 (2025)
-
[31]
Erickson, A.et al.Spatially resolved clonal copy number alterations in benign and malignant tissue.Nature608, 360–367 (2022)
2022
-
[32]
D.et al.Image analysis with deep learning to predict breast cancer grade, er status, histologic subtype, and intrinsic subtype.NPJ breast cancer4, 30 (2018)
Couture, H. D.et al.Image analysis with deep learning to predict breast cancer grade, er status, histologic subtype, and intrinsic subtype.NPJ breast cancer4, 30 (2018)
2018
-
[33]
Valieris, R.et al.Weakly-supervised deep learning models enable her2-low prediction from h &e stained slides.Breast Cancer Res.26, 124 (2024)
2024
-
[34]
H.et al.Estrogen and progesterone receptor testing in breast cancer: Asco/cap guideline update.J
Allison, K. H.et al.Estrogen and progesterone receptor testing in breast cancer: Asco/cap guideline update.J. Clin. Oncol. 38, 1346–1366 (2020)
2020
-
[35]
Wolff, A. C.et al.Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of american pathologists clinical practice guideline focused update.Arch. pathology & laboratory medicine142, 1364–1382 (2018)
2018
-
[36]
D.et al.The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification.J
Travis, W. D.et al.The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification.J. thoracic oncology10, 1243–1260 (2015). 28/51
2015
-
[37]
thoracic oncology 14, 377–407 (2019)
Yatabe, Y .et al.Best practices recommendations for diagnostic immunohistochemistry in lung cancer.J. thoracic oncology 14, 377–407 (2019)
2019
-
[38]
A.et al.p40 ( δnp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma.Mod
Bishop, J. A.et al.p40 ( δnp63) is superior to p63 for the diagnosis of pulmonary squamous cell carcinoma.Mod. pathology25, 405–415 (2012)
2012
-
[39]
Mosele, F.et al.Recommendations for the use of next-generation sequencing (ngs) for patients with metastatic cancers: a report from the esmo precision medicine working group.Annals Oncol.31, 1491–1505 (2020)
2020
-
[40]
medicine24, 1559–1567 (2018)
Coudray, N.et al.Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning.Nat. medicine24, 1559–1567 (2018)
2018
-
[41]
T.et al.Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade.Science357, 409–413 (2017)
Le, D. T.et al.Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade.Science357, 409–413 (2017)
2017
-
[42]
The Lancet Oncol.21, 1353–1365 (2020)
Marabelle, A.et al.Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 keynote-158 study. The Lancet Oncol.21, 1353–1365 (2020)
2020
-
[43]
R.et al.Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden
Chalmers, Z. R.et al.Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome medicine9, 34 (2017)
2017
-
[44]
McGrail, D.et al.High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types.Annals Oncol.32, 661–672 (2021)
2021
-
[45]
13, B. . W. H. . H. M. S. C. L. . . P. P. J. . K. R., data analysis: Baylor College of Medicine Creighton Chad J. 22 23 Donehower Lawrence A. 22 23 24 25, G., for Systems Biology Reynolds Sheila 31 Kreisberg Richard B. 31 Bernard Brady 31 Bressler Ryan 31 Erkkila Timo 32 Lin Jake 31 Thorsson Vesteinn 31 Zhang Wei 33 Shmulevich Ilya 31, I.et al. Comprehens...
2012
-
[46]
S.et al.Supervised risk predictor of breast cancer based on intrinsic subtypes.J
Parker, J. S.et al.Supervised risk predictor of breast cancer based on intrinsic subtypes.J. clinical oncology27, 1160–1167 (2009). 57.Guinney, J.et al.The consensus molecular subtypes of colorectal cancer.Nat. medicine21, 1350–1356 (2015). 58.Sanchez-Vega, F.et al.Oncogenic signaling pathways in the cancer genome atlas.Cell173, 321–337 (2018)
2009
-
[47]
& Jaffee, E
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to pd-1 inhibition.New Engl. J. Medicine377, 2500–2501 (2017)
2017
-
[48]
61.Kleppe, A.et al.Designing deep learning studies in cancer diagnostics.Nat
Litchfield, K.et al.Meta-analysis of tumor-and t cell-intrinsic mechanisms of sensitization to checkpoint inhibition.Cell 184, 596–614 (2021). 61.Kleppe, A.et al.Designing deep learning studies in cancer diagnostics.Nat. Rev. Cancer21, 199–211 (2021)
2021
-
[49]
& V oet, T
Vandereyken, K., Sifrim, A., Thienpont, B. & V oet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet.24, 494–515 (2023)
2023
-
[50]
& Hochreiter, S
Klambauer, G., Unterthiner, T., Mayr, A. & Hochreiter, S. Self-normalizing neural networks.Adv. neural information processing systems30(2017). 64.Vaswani, A.et al.Attention is all you need.Adv. neural information processing systems30(2017)
2017
-
[51]
Representation Learning with Contrastive Predictive Coding
Oord, A. v. d., Li, Y . & Vinyals, O. Representation learning with contrastive predictive coding.arXiv preprint arXiv:1807.03748(2018). 29/51 Extended Data Extended Data Table 1.Dataset details for spatial gene expression prediction.There are a total of 8 datasets across 8 distinct cancer types. The table provides the overview of the oncology cohorts, the...
work page internal anchor Pith review Pith/arXiv arXiv 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.