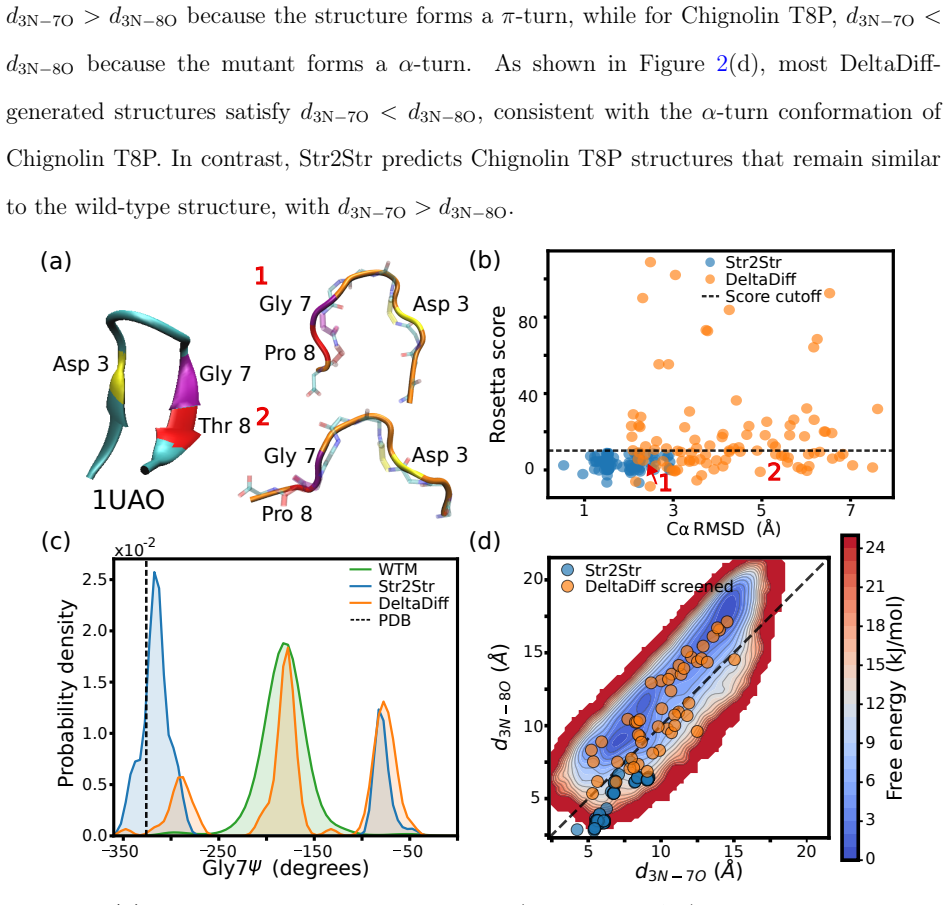
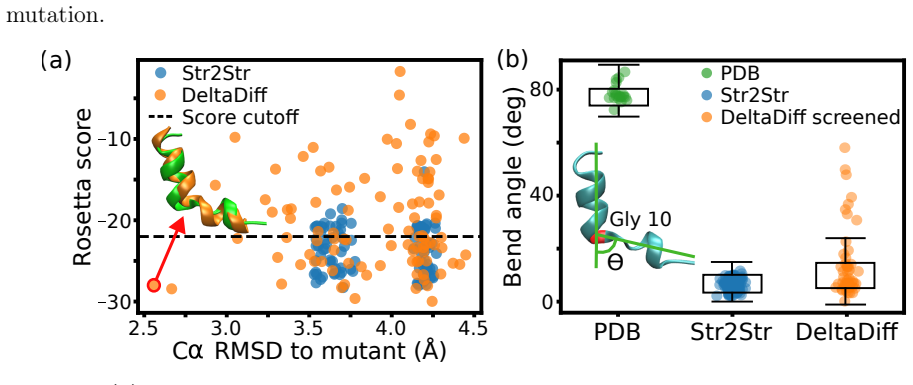
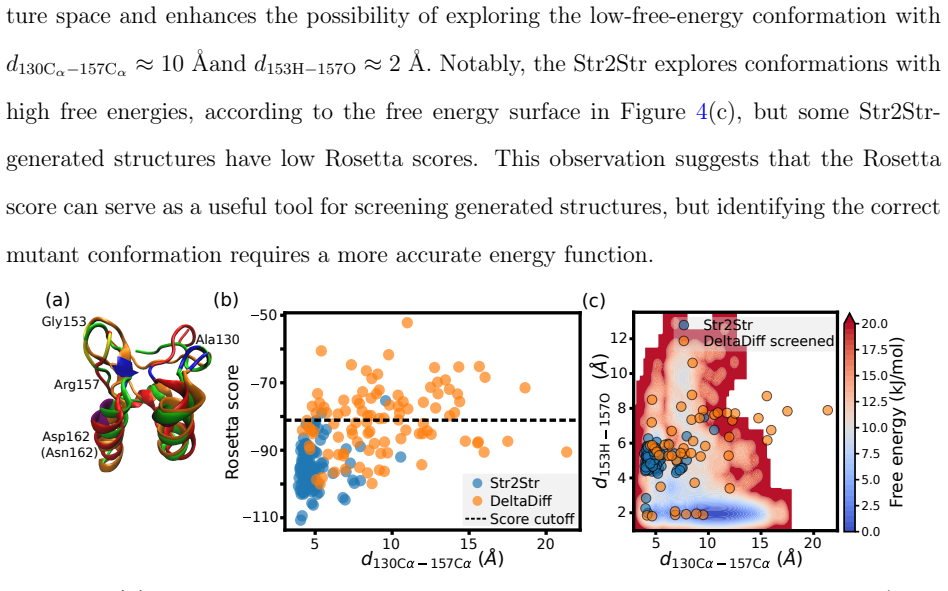
DeltaDiff: Training-Free, Physics-Guided Machine Learning for Predicting Mutant Protein Structures
Pith reviewed 2026-06-28 04:13 UTC · model grok-4.3
The pith
DeltaDiff adds mutation-aware physical guidance to a pre-trained diffusion model to predict mutant protein structures without retraining.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
DeltaDiff is a physics-guided inference framework that incorporates mutation-aware physical guidance into a baseline diffusion model. Evaluated on Chignolin T8P, Novispirin G-10, and BBL D162N, systems that each exhibit nonlocal structural changes, DeltaDiff captures the mutation-induced conformational shifts without any retraining or fine-tuning of the baseline model.
What carries the argument
DeltaDiff, the inference-time framework that augments a baseline diffusion model with mutation-aware physical guidance.
If this is right
- Mutant structure prediction becomes feasible at a fraction of the cost of conventional modeling or experimental methods.
- Rational design of mutants can proceed by generating candidate structures directly from sequence changes.
- The same guidance approach may apply to other single-site or few-site sequence variants that induce conformational shifts.
- Training data collection and model retraining are no longer required for each new mutant of interest.
Where Pith is reading between the lines
- Physical priors supplied at inference time can substitute for task-specific training data in structure-prediction networks.
- The method may generalize to other biomolecules where small sequence edits produce large structural effects.
- Integration with additional pre-trained models could further lower the barrier to exploring mutational landscapes.
- The approach highlights a route to make existing large models more adaptable without additional compute for retraining.
Load-bearing premise
A pre-trained diffusion model can be steered at inference time by physical rules that encode mutation effects and thereby recover nonlocal structural rearrangements that the model was never explicitly trained to produce.
What would settle it
High-resolution experimental structures of the three tested mutants that deviate substantially from the DeltaDiff predictions in the regions reported to change conformation.
Figures

read the original abstract
Determining mutant protein structures is critical for understanding the mechanistic roles of mutations in biochemical processes. However, experimental characterization and conventional theoretical modeling are often expensive and time-consuming. Recent advances in machine learning provide new opportunities to efficiently predict protein structures from primary sequences. Nevertheless, applying these models to proteins with single-site or few-site mutations remains challenging because mutant sequences are often highly similar to their wild-type counterparts. Here, we introduce DeltaDiff, a physics-guided inference framework for mutant-structure generation that incorporates mutation-aware physical guidance into a baseline diffusion model. We evaluate DeltaDiff on three representative systems: Chignolin T8P, Novispirin G-10, and BBL D162N. All three examples involve nonlocal structural changes, making accurate mutant-structure prediction challenging. DeltaDiff captures key mutation-induced conformational changes without requiring retraining or fine-tuning of the baseline model. These results establish a foundation for efficient mutant-structure prediction at a fraction of the cost of conventional methods, facilitating rational mutant design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces DeltaDiff, a training-free physics-guided inference framework that augments a baseline diffusion model with mutation-aware physical guidance during sampling to predict structures of mutant proteins. It is evaluated on three systems exhibiting nonlocal conformational changes (Chignolin T8P, Novispirin G-10, BBL D162N) and claims that the guidance enables capture of key mutation-induced changes without retraining or fine-tuning the baseline model, at lower cost than conventional approaches.
Significance. If the central claim holds with quantitative support, the training-free aspect would be a notable strength for efficient mutant-structure prediction in computational biophysics and protein design, avoiding the expense of retraining large diffusion models while leveraging existing ones. The focus on nonlocal changes addresses a known challenge in sequence-similar mutants.
major comments (2)
- [Abstract / Results] The provided description (including abstract) contains no quantitative metrics (e.g., RMSD, GDT-TS, or contact-map accuracy) or direct comparisons to the unaugmented baseline diffusion model or other mutant-prediction methods. This is load-bearing for the claim that the guidance accurately captures nonlocal changes on the three systems.
- [Methods] The implementation of 'mutation-aware physical guidance' is not specified with equations or pseudocode (e.g., how mutation effects enter the score function or energy terms during inference). Without this, it is not possible to verify that the approach is truly training-free and non-circular for nonlocal effects.
minor comments (2)
- Clarify the baseline diffusion model used (architecture, training data) and any hyper-parameters of the guidance term.
- Add a table or figure summarizing structural metrics for wild-type vs. mutant predictions across the three systems.
Simulated Author's Rebuttal
We thank the referee for the constructive report and the recommendation for major revision. We agree that quantitative metrics and explicit methodological details are necessary to support the central claims. Below we respond point-by-point and indicate the revisions that will be made.
read point-by-point responses
-
Referee: [Abstract / Results] The provided description (including abstract) contains no quantitative metrics (e.g., RMSD, GDT-TS, or contact-map accuracy) or direct comparisons to the unaugmented baseline diffusion model or other mutant-prediction methods. This is load-bearing for the claim that the guidance accurately captures nonlocal changes on the three systems.
Authors: We agree that quantitative support is essential. The current manuscript reports only qualitative observations of conformational changes on the three systems. In the revised version we will add RMSD, GDT-TS, and contact-map accuracy metrics for both DeltaDiff and the unaugmented baseline diffusion model on all three test cases (Chignolin T8P, Novispirin G-10, BBL D162N). We will also include a direct comparison table and, where feasible, reference to existing mutant-prediction methods. These additions will be placed in a new Results subsection and summarized in the abstract. revision: yes
-
Referee: [Methods] The implementation of 'mutation-aware physical guidance' is not specified with equations or pseudocode (e.g., how mutation effects enter the score function or energy terms during inference). Without this, it is not possible to verify that the approach is truly training-free and non-circular for nonlocal effects.
Authors: We acknowledge that the Methods section currently describes the guidance at a high level without explicit equations. In the revision we will insert a dedicated subsection containing the precise formulation: the modified score function s_θ(x_t, t; ΔE_mut) where the mutation-induced energy term ΔE_mut is added to the denoising step, together with pseudocode for the guided sampling loop. This will make clear that no model parameters are updated and that the guidance remains non-circular because it uses only the fixed baseline model plus a physics-based energy evaluator. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper presents DeltaDiff as a training-free inference framework that augments an existing baseline diffusion model with mutation-aware physical guidance. No equations, derivations, fitted parameters, or self-citation chains are described in the provided abstract or claim summary. The central claim reduces to an empirical demonstration on three systems rather than any mathematical reduction of a prediction to its own inputs. The method is therefore self-contained against external benchmarks with no load-bearing circular steps.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Plos one , volume=
Using AlphaFold to predict the impact of single mutations on protein stability and function , author=. Plos one , volume=. 2023 , publisher=
2023
-
[2]
Journal of Chemical Information and Modeling , volume=
Evaluation of AlphaFold 3’s protein--protein complexes for predicting binding free energy changes upon mutation , author=. Journal of Chemical Information and Modeling , volume=. 2024 , publisher=
2024
-
[3]
Computers in biology and medicine , volume=
Integration of persistent Laplacian and pre-trained transformer for protein solubility changes upon mutation , author=. Computers in biology and medicine , volume=. 2024 , publisher=
2024
-
[4]
Nature communications , volume=
Machine learning coarse-grained potentials of protein thermodynamics , author=. Nature communications , volume=. 2023 , publisher=
2023
-
[5]
Proceedings of the National Academy of Sciences , volume=
Protein language models trained on biophysical dynamics inform mutation effects , author=. Proceedings of the National Academy of Sciences , volume=. 2026 , publisher=
2026
-
[6]
Science , volume=
Evolutionary-scale prediction of atomic-level protein structure with a language model , author=. Science , volume=. 2023 , publisher=
2023
-
[7]
Science , volume=
Scalable emulation of protein equilibrium ensembles with generative deep learning , author=. Science , volume=. 2025 , publisher=
2025
-
[8]
Nature , volume=
De novo design of protein structure and function with RFdiffusion , author=. Nature , volume=. 2023 , publisher=
2023
-
[9]
International Conference on Learning Representations , volume=
Str2str: A score-based framework for zero-shot protein conformation sampling , author=. International Conference on Learning Representations , volume=
-
[10]
bioRxiv , pages=
Rapid sequence-based screening of structure-disrupting protein mutations , author=. bioRxiv , pages=. 2026 , publisher=
2026
-
[11]
Structure , volume=
10 residue folded peptide designed by segment statistics , author=. Structure , volume=. 2004 , publisher=
2004
-
[12]
RSC advances , volume=
Mutation-induced change in chignolin stability from -turn to -turn , author=. RSC advances , volume=. 2020 , publisher=
2020
-
[13]
Deep mutational scanning: a new style of protein science , author =. Nature Methods , volume =. 2014 , publisher =. doi:10.1038/nmeth.3027 , url =
-
[14]
Nature Reviews Molecular Cell Biology , volume =
Exploring protein fitness landscapes by directed evolution , author =. Nature Reviews Molecular Cell Biology , volume =. 2009 , publisher =. doi:10.1038/nrm2805 , url =
-
[15]
Current Opinion in Structural Biology , volume =
Stability effects of mutations and protein evolvability , author =. Current Opinion in Structural Biology , volume =. 2009 , publisher =. doi:10.1016/j.sbi.2009.08.003 , url =
-
[16]
Current Opinion in Structural Biology , volume =
Allostery without a conformational change? Revisiting the paradigm , author =. Current Opinion in Structural Biology , volume =. 2015 , publisher =. doi:10.1016/j.sbi.2014.11.005 , url =
-
[17]
Annual Review of Biophysics , volume =
The Role of Conformational Dynamics and Allostery in Modulating Protein Evolution , author =. Annual Review of Biophysics , volume =. 2020 , publisher =. doi:10.1146/annurev-biophys-052118-115517 , url =
-
[18]
Highly Accurate Protein Structure Prediction with
Highly accurate protein structure prediction with AlphaFold , author =. Nature , volume =. 2021 , publisher =. doi:10.1038/s41586-021-03819-2 , url =
-
[19]
Accurate prediction of protein structures and interactions using a three-track neural network , author =. Science , volume =. 2021 , publisher =. doi:10.1126/science.abj8754 , url =
-
[20]
Accurate structure prediction of biomolecular interactions with AlphaFold 3 , author =. Nature , volume =. 2024 , publisher =. doi:10.1038/s41586-024-07487-w , url =
-
[21]
2020 , eprint =
Score-Based Generative Modeling through Stochastic Differential Equations , author =. 2020 , eprint =
2020
-
[22]
Nature Computational Science , volume =
Score-based generative modeling for de novo protein design , author =. Nature Computational Science , volume =. 2023 , publisher =. doi:10.1038/s43588-023-00440-3 , url =
-
[23]
Lawrence Zitnick, Jerry Ma, and Rob Fergus
Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences , author =. Proceedings of the National Academy of Sciences , volume =. 2021 , publisher =. doi:10.1073/pnas.2016239118 , url =
-
[24]
Language models enable zero-shot prediction of the effects of mutations on protein function , author =. 2021 , publisher =. doi:10.1101/2021.07.09.450648 , url =
-
[25]
MSA Transformer , author =. 2021 , publisher =. doi:10.1101/2021.02.12.430858 , url =
-
[26]
2022 , eprint =
Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval , author =. 2022 , eprint =
2022
-
[27]
Genome-wide prediction of disease variant effects with a deep protein language model , author =. Nature Genetics , volume =. 2023 , publisher =. doi:10.1038/s41588-023-01465-0 , url =
-
[28]
Nature Structural &; Molecular Biology , volume =
Can AlphaFold2 predict the impact of missense mutations on structure? , author =. Nature Structural &; Molecular Biology , volume =. 2022 , publisher =. doi:10.1038/s41594-021-00714-2 , url =
-
[29]
Nature Chemical Biology , volume =
The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins , author =. Nature Chemical Biology , volume =. 2024 , publisher =. doi:10.1038/s41589-024-01638-w , url =
-
[30]
Physical Review Letters , volume =
AlphaFold2 Can Predict Single-Mutation Effects , author =. Physical Review Letters , volume =. 2023 , publisher =. doi:10.1103/PhysRevLett.131.218401 , url =
-
[31]
2021 , eprint=
Diffusion Models Beat GANs on Image Synthesis , author=. 2021 , eprint=
2021
-
[32]
Machine Learning Force Fields , author =. Chemical Reviews , volume =. 2021 , publisher =. doi:10.1021/acs.chemrev.0c01111 , url =
-
[33]
E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials , author =. Nature Communications , volume =. 2022 , publisher =. doi:10.1038/s41467-022-29939-5 , url =
-
[34]
2022 , eprint=
TorchMD-NET: Equivariant Transformers for Neural Network based Molecular Potentials , author=. 2022 , eprint=
2022
-
[35]
Nature Communications , volume=
Engineering protein-based therapeutics through structural and chemical design , author=. Nature Communications , volume=. 2023 , doi=
2023
-
[36]
Protein engineering , volume=
Impact of single-residue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides , author=. Protein engineering , volume=. 2002 , publisher=
2002
-
[37]
Journal of molecular biology , volume=
The folding mechanism of BBL: Plasticity of transition-state structure observed within an ultrafast folding protein family , author=. Journal of molecular biology , volume=. 2009 , publisher=
2009
-
[38]
Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface , author =. Science , volume =. 1985 , publisher =. doi:10.1126/science.4001944 , url =
-
[39]
Proceedings of the National Academy of Sciences , volume =
Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence , author =. Proceedings of the National Academy of Sciences , volume =. 2010 , publisher =. doi:10.1073/pnas.1004290107 , url =
-
[40]
Nature Structural Biology , volume =
The way to NMR structures of proteins , author =. Nature Structural Biology , volume =. 2001 , publisher =. doi:10.1038/nsb1101-923 , url =
-
[41]
The Resolution Revolution , author =. Science , volume =. 2014 , publisher =. doi:10.1126/science.1251652 , url =
-
[42]
Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Models , author=
-
[43]
Using circular dichroism spectra to estimate protein secondary structure , author =. Nature Protocols , volume =. 2006 , publisher =. doi:10.1038/nprot.2006.202 , url =
-
[44]
Annual Review of Biophysics , volume =
Biomolecular Simulation: A Computational Microscope for Molecular Biology , author =. Annual Review of Biophysics , volume =. 2012 , publisher =. doi:10.1146/annurev-biophys-042910-155245 , url =
-
[45]
Accounts of Chemical Research , volume =
Molecular Dynamics Simulations of Biomolecules , author =. Accounts of Chemical Research , volume =. 2002 , publisher =. doi:10.1021/ar020082r , url =
-
[46]
Protein complex prediction with AlphaFold-Multimer , author =. 2021 , publisher =. doi:10.1101/2021.10.04.463034 , url =
-
[47]
Dictionary of protein secondary structure: Pattern recognition of hydrogen‐bonded and geometrical features , author =. Biopolymers , volume =. 1983 , publisher =. doi:10.1002/bip.360221211 , url =
-
[48]
ExEnDiff: An Experiment-Guided Diffusion Model for Protein Conformational Ensemble Generation , author =. PRX Life , volume =. 2025 , publisher =. doi:10.1103/PRXLife.3.023013 , url =
-
[49]
The Journal of Physical Chemistry Letters , volume =
Extrapolating Foundation Generative Models with Physics: A Case Study of Exploring Peptide Conformations under Protein–Environment Interactions , author =. The Journal of Physical Chemistry Letters , volume =. 2025 , publisher =. doi:10.1021/acs.jpclett.5c02567 , url =
-
[50]
bioRxiv , year=
ESMAdam: A Plug-and-Play All-Purpose Protein Ensemble Generator , author=. bioRxiv , year=
-
[51]
Nature Reviews Microbiology , volume =
Molecular mechanisms of antibiotic resistance , author =. Nature Reviews Microbiology , volume =. 2014 , publisher =. doi:10.1038/nrmicro3380 , url =
-
[52]
Widespread Macromolecular Interaction Perturbations in Human Genetic Disorders , author =. Cell , volume =. 2015 , publisher =. doi:10.1016/j.cell.2015.04.013 , url =
-
[53]
Nature Reviews Chemistry , volume =
Loop dynamics and the evolution of enzyme activity , author =. Nature Reviews Chemistry , volume =. 2023 , publisher =. doi:10.1038/s41570-023-00495-w , url =
-
[54]
Proceedings of the National Academy of Sciences , volume =
Interconversion between two unrelated protein folds in the lymphotactin native state , author =. Proceedings of the National Academy of Sciences , volume =. 2008 , publisher =. doi:10.1073/pnas.0709518105 , url =
-
[55]
Proceedings of the National Academy of Sciences , volume =
Reversible switching between two common protein folds in a designed system using only temperature , author =. Proceedings of the National Academy of Sciences , volume =. 2023 , publisher =. doi:10.1073/pnas.2215418120 , url =
-
[56]
Nature Reviews Microbiology , volume =
Molecular mechanisms of antibiotic resistance revisited , author =. Nature Reviews Microbiology , volume =. 2022 , publisher =. doi:10.1038/s41579-022-00820-y , url =
-
[57]
Nature Reviews Molecular Cell Biology , volume =
Structural and functional constraints in the evolution of protein families , author =. Nature Reviews Molecular Cell Biology , volume =. 2009 , publisher =. doi:10.1038/nrm2762 , url =
-
[58]
High-resolution mapping of protein sequence-function relationships , author =. Nature Methods , volume =. 2010 , publisher =. doi:10.1038/nmeth.1492 , url =
-
[59]
Nucleic Acids Research , volume =
The Protein Data Bank , author =. Nucleic Acids Research , volume =. 2000 , publisher =. doi:10.1093/nar/28.1.235 , url =
-
[60]
Nucleic Acids Research , volume =
AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models , author =. Nucleic Acids Research , volume =. 2021 , publisher =. doi:10.1093/nar/gkab1061 , url =
-
[61]
Current Opinion in Structural Biology , volume =
Proteins that switch folds , author =. Current Opinion in Structural Biology , volume =. 2010 , publisher =. doi:10.1016/j.sbi.2010.06.002 , url =
-
[62]
Mutational Tipping Points for Switching Protein Folds and Functions , author =. Structure , volume =. 2012 , publisher =. doi:10.1016/j.str.2011.11.018 , url =
-
[63]
2022 , eprint =
Classifier-Free Diffusion Guidance , author =. 2022 , eprint =
2022
-
[64]
2020 , eprint =
Denoising Diffusion Probabilistic Models , author =. 2020 , eprint =
2020
-
[65]
Proceedings of the National Academy of Sciences , volume =
An end-to-end deep learning method for protein side-chain packing and inverse folding , author =. Proceedings of the National Academy of Sciences , volume =. 2023 , publisher =. doi:10.1073/pnas.2216438120 , url =
-
[66]
PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta , author =. Bioinformatics , volume =. 2010 , publisher =. doi:10.1093/bioinformatics/btq007 , url =
-
[67]
Journal of Chemical Theory and Computation , volume =
The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design , author =. Journal of Chemical Theory and Computation , volume =. 2017 , publisher =. doi:10.1021/acs.jctc.7b00125 , url =
-
[68]
Folding free‐energy landscape of a 10‐residue mini‐protein, chignolin , author =. FEBS Letters , volume =. 2006 , publisher =. doi:10.1016/j.febslet.2006.05.015 , url =
-
[69]
Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method
Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method , author =. Physical Review Letters , volume =. 2008 , publisher =. doi:10.1103/PhysRevLett.100.020603 , url =
-
[70]
2023 , eprint =
Steered Diffusion: A Generalized Framework for Plug-and-Play Conditional Image Synthesis , author =. 2023 , eprint =
2023
-
[71]
Nature Reviews Genetics , volume =
Missense meanderings in sequence space: a biophysical view of protein evolution , author =. Nature Reviews Genetics , volume =. 2005 , publisher =. doi:10.1038/nrg1672 , url =
-
[72]
Nature Reviews Genetics , volume =
Mutational effects and the evolution of new protein functions , author =. Nature Reviews Genetics , volume =. 2010 , publisher =. doi:10.1038/nrg2808 , url =
-
[73]
Mega-scale experimental analysis of protein folding stability in biology and design , author =. Nature , volume =. 2023 , publisher =. doi:10.1038/s41586-023-06328-6 , url =
-
[74]
Accurate proteome-wide missense variant effect prediction with AlphaMissense , author =. Science , volume =. 2023 , publisher =. doi:10.1126/science.adg7492 , url =
-
[75]
Disease variant prediction with deep generative models of evolutionary data , author =. Nature , volume =. 2021 , publisher =. doi:10.1038/s41586-021-04043-8 , url =
-
[76]
Deep generative models of genetic variation capture the effects of mutations , author =. Nature Methods , volume =. 2018 , publisher =. doi:10.1038/s41592-018-0138-4 , url =
-
[77]
Nature Biotechnology , volume =
Mutation effects predicted from sequence co-variation , author =. Nature Biotechnology , volume =. 2017 , publisher =. doi:10.1038/nbt.3769 , url =
-
[78]
Dynamic personalities of proteins , author =. Nature , volume =. 2007 , publisher =. doi:10.1038/nature06522 , url =
-
[79]
The Energy Landscapes and Motions of Proteins , author =. Science , volume =. 1991 , publisher =. doi:10.1126/science.1749933 , url =
-
[80]
The Protein-Folding Problem, 50 Years On , author =. Science , volume =. 2012 , publisher =. doi:10.1126/science.1219021 , url =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.